
Abstract
Aim
Migraine pain is thought to result from activation of meningeal nociceptors that might involve dural mast cell degranulation and release of proteases and pronociceptive mediators. Tryptase, the most abundant dural mast cell protease, has been demonstrated to stimulate dural mast cells, as well as trigeminal nociceptors by activating the protease activated receptor 2. Mast cell or neuronal protease activated receptors 2 may therefore represent a novel target for migraine treatment. In this study, we characterized and evaluated a novel protease activated receptor 2 monoclonal antibody as a preventive anti-migraine pain therapy in preclinical models.
Methods
Flow cytometry, immunocytochemistry, calcium imaging, Homogeneous Time Resolved Technology (HTRF) epitope competition assay and serum pharmacokinetic (PK) assay in rats were performed to confirm the activity, specificity and in vivo stability of PAR650097, a novel anti- protease activated receptor 2 monoclonal antibody. In vivo assessment was performed in female C57BL/6J mice by evaluation of PAR650097 in preventing cutaneous allodynia elicited by (a) supradural injection of the protease activated receptor 2 agonist, Ser-Leu-Ile-Gly-Arg-Leu-amide trifluoroacetate (SLIGRL), or calcitonin gene-related (CGRP) peptide, and (b) induction of latent sensitization by priming with three daily episodes of restraint stress followed by challenge with a subthreshold inhalational exposure to umbellulone (UMB), a transient receptor potential ankyrin 1 (TRPA1) agonist. PAR650097 was administered as a pretreatment prior to the first restraint stress, umbellulone exposure, SLIGRL or calcitonin gene-related peptide injection. Additionally, fremanezumab, a calcitonin gene-related peptide antibody was administered as pre-treatment prior to supradural administration of calcitonin gene-related peptide or SLIGRL.
Results
In vitro, PAR650097 demonstrated rapid interaction with protease activated receptor 2, enabling it to fully inhibit protease-induced protease activated receptor 2 activation, in human and mouse cells, with high potency. Furthermore, PAR650097 was highly selective for protease activated receptor 2, demonstrating no affinity for protease activated receptor 1 protein and no functional effect on the activation of cellular protease activated receptor 1 with thrombin. In addition, PAR650097 had an acceptable PK profile, compatible with testing the effects of selective protease activated receptor 2 inhibition in vivo. In vivo, PAR650097 blocked cutaneous allodynia induced by either supradural SLIGRL or calcitonin gene-related peptide. Fremanezumab abolished cutaneous allodynia induced by supradural CGRP, and partially attenuated cutaneous allodynia induced by SLIGRL. Administration of PAR650097, before the first restraint stress episode, did not prevent the acute stress-induced cutaneous allodynia or restraint stress priming revealed by cutaneous allodynia induced by inhalational umbellulone. In contrast, PAR650097 prevented expression of cutaneous allodynia when given before the umbellulone challenge in restraint stress-primed animals.
Conclusion
PAR650097 specifically inhibits endogenously expressed protease activated receptor 2 in human and mouse cells with high potency. This antibody has an acceptable PK profile in rodents and effectively blocked SLIGR-induced cutaneous allodynia. PAR650097 additionally prevented cutaneous allodynia induced by supradural calcitonin gene-related peptide, indicating that the protease activated receptor 2 receptor is a downstream consequence of calcitonin gene-related peptide actions. Fremanezumab effectively blocked calcitonin gene-related peptide-induced cutaneous allodynia and only partially reduced cutaneous allodynia induced by a protease activated receptor 2 activator, suggesting both calcitonin gene-related peptide-dependent and -independent mechanisms in promoting migraine pain. While PAR650097 did not prevent stress-induced cutaneous allodynia or priming, it effectively prevented cutaneous allodynia induced by a TRPA1 agonist in animals with latent sensitization. Activation of protease activated receptor 2, therefore, contributes to both calcitonin gene-related peptide-dependent and -independent mechanisms in promoting migraine-like pain. Therapeutic targeting of protease activated receptor 2 receptors may represent an anti-migraine pain strategy with a potentially broad efficacy profile.
Introduction
Migraine represents one of the most common neurological disorders, and leads to severe global, social and occupational impairments. Migraine is commonly characterized by different phases, intermittent pain that occurs in the absence of injury, and trigeminovascular system activation that is associated with release of pronociceptive mediators including calcitonin-gene related peptide (CGRP) (1–4). In spite of numerous medications available for acute and preventive treatment of migraine, many people with migraine remain poorly managed, likely due to the complex interplay of genetic and biological features of the disorder (2–6). A deeper understanding of the mechanisms underlying migraine pathophysiology may lead to the discovery of new mechanistic strategies to improve therapeutic outcomes.
Current understanding suggests that migraine pain ultimately requires activation of nociceptors in the cranial meninges (2–4). This process likely involves dural immune mast cell (MC) degranulation, which leads to secretion of inflammatory mediators, cytokines, chemokines and proteases, all of which may promote activation of meningeal nociceptors (7–10). Tryptase, the most abundant MC protease, has been demonstrated to stimulate MC, as well as trigeminal nociceptors, by activating the protease activated receptor 2 (PAR2) (7–9). PAR2 is a receptor belonging to the G protein-coupled Ca2+-mobilizing PAR1–4 receptor family (11), which has a unique activation mechanism through a so-called “tethered ligand”, where cleavage of a site on the extracellular N-terminal domain generates the endogenous ligand responsible for cell signaling (11,12). PAR2 receptors are also activated by the exogenous proteases or non-proteolytically generated peptides, such as SLIGRL, a compound that mimics the final amino acids of the tethered endogenous ligands (11,12). Due to the wide expression throughout multiple body systems, both MC and neuronal PAR2 receptors have been implicated in numerous pathological pain states, including in migraine (11–15).
We recently characterized and developed an injury-free model using the “two-hit” priming strategy, which induces vulnerability, termed latent sensitization (LS), allowing the study of mechanisms relevant to migraine-like pain and the evaluation of novel preventive or acute treatments (16). Repeated stress episodes were used as the “first hit” to produce a transient cutaneous allodynia (CA) and LS (i.e. priming) that was revealed by challenge with subthreshold inhalational exposure to umbellulone (UMB), a TRPA1 agonist, as the “second hit” (16). Recent studies have revealed that supradural injection of CGRP promotes migraine-like pain behaviors, potentially mimicking the action of endogenous CGRP peptide in meningeal neurons (17). Herein, we characterized a specific PAR2 monoclonal antibody (mAb), PAR650097, with respective receptor binding, function and pharmacokinetic (PK) studies and subsequently evaluated its effects in preventing migraine-like pain behaviors.
Materials and methods
Animals
Behavioral experiments were conducted using 6-week-old female C57BL/6J mice (Jackson Labs, Sacramento, CA, USA). Animals were housed in a 12 h light/dark cycle (lights on 7am to 7pm), climate- and humidity-controlled environment with free access to food and water in the University of Arizona animal facility. A total of 216 animals were used in these studies with 6–17 animals per group size for evaluation of behavior. Sample size was determined using GPower 3.1 software, with an established significance level of p < 0.05. All experimental procedures were performed in accordance with the ARRIVE guidelines, the ethical guidelines of the International Association for the Study of Pain regulations on animal welfare and the National Institutes of Health guidelines for the care and use of laboratory animals. The experimental procedures have been previously approved by the Institutional Animal Care and Use Committee of the University of Arizona. Mice were randomly divided into control and experimenters were blinded to treatments and behavioral measurements. Pharmacokinetic studies were performed in male CD rats that were obtained from Charles River (Cambridge, UK), under procedures and protocols previously approved by Huntingdon Life Sciences and AstraZeneca.
Drugs
SLIGRL (Sigma, St. Louis, MO, USA) was dissolved in synthetic interstitial fluid (SIF, pH 7.4). The SIF contained 10 mM HEPES, 5 mM KCl, 135 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM glucose (all from Sigma, St. Louis, MO, USA). The humanized PAR2 mAb (PAR650097, AstraZeneca, Cambridge, UK) and isotype control protein (R347 HULGg1 TM, AstraZeneca, Cambridge, UK) were diluted in phosphate-buffered saline (PBS). Umbellulone (UMB; AdipoGen, San Diego, CA, USA) was prepared as a stock solution of 0.1 M in 100% dimethyl sulfoxide (DMSO) and freshly diluted to 0.01 M with PBS. CGRP (Bachem, Torrance, CA, USA) was dissolved in deionized water. The humanized anti-CGRP mAb fremanezumab (i.e. AJOVY®) was purchased from the University of Arizona hospital pharmacy and was diluted in PBS. UMB was delivered by the inhalational route, while PAR2 antibody and control protein were administered intraperitoneally (i.p.) at 30 or 100 mg/kg, anti-CGRP mAb was given i.p. at 30 mg/kg; SLIGRL at 1, 3, 10 and 30 µg/5 µL, and CGRP at 1 pg/5 µL were delivered by supradural administration. Controls received the administration of the respective vehicle.
Generation of recombinant human PAR2 and PAR1 proteins
The human PAR2 (Proteinase-Activated Receptor 2) constructs comprising extracellular residues 1-75 were designed with N-terminal AviTag™ (Avidity LLC, Aurora, CO, USA) and C-terminal Flag and poly Histidine tags and cloned into the vector pDEST12.2 OriP FH (Life Technologies, Carlsbad, CA, USA). The human PAR1 (Proteinase-Activated Receptor 1) construct comprising extracellular residues 1-102 was designed with C-terminal Flag and poly Histidine tags and cloned into the vector pDEST12.2 OriP FH (Life Technologies, Carlsbad, CA, USA). The constructs were expressed in HEK293 cells and purified from the media using standard affinity and size exclusion chromatography purification. To generate biotinylated proteins, the AviTag™ was biotinylated enzymatically according to the manufacturer’s instructions.
Isolation of anti-PAR2 mAb PAR650097
PAR650097 was isolated from a large phage library displaying human single-chain variable fragment (scFv) (18–20) by carrying out panning and solution phase selections on recombinant human PAR2. This was followed by in vitro affinity maturation via targeted and random mutagenesis of the complementarity determining regions (CDRs) using phage display essentially, as previously described (21).
Reformatting of scFv to IgG
PAR650097 was converted from scFv to immunoglobulin G (IgG) format by subcloning the VH and VL domains into human IgG heavy chain and light chain expression vectors based on those originally described by Persic and colleagues (22) with an additional OriP element engineered into each. The plasmids were co-transfected into HEK293/EBNA mammalian cells for expression and IgG proteins purified from the culture medium using Protein A chromatography.
Affinity analysis measured by surface plasmon resonance (SPR)
The BIAcore 2000 (GE Healthcare, Little Chalfont, UK) biosensor instrument was used to assess the kinetic parameters of the interaction between the PAR650097 antibody and recombinant human PAR2 His Flag. The affinity of binding between each sample and the analyte was calculated using assays in which a constant amount of IgG was captured by affinity for Protein G’ that had been covalently coupled by amine linkage to a proprietary CM5 chip surface. Human PAR2 His Flag prepared in HEPES-buffered saline with Ethylenediaminetetraacetic acid and surfactant P (HBS-EP) buffer (BIAcore AB), at a range of concentrations, between 200 nM and 6.25 nM, were passed over the sensor chip surface. The surface was regenerated using 10 mM Glycine, pH 1.75 between each injection of antibody. The resulting sensorgrams were evaluated using BIA evaluation 4.1 software and fitted to a 1:1 Langmuir binding model, to provide the relative binding data. Control experiments were carried out to determine that there was minimal mass transport; the interaction was unaffected by the choice of buffer, and the interaction was truly a 1:1 binding event.
Flow cytometry
1321N1 parental cell line (lacking PAR2 expression)(23) or 1321N1 cells stably expressing human PAR2 (1321N1-hPAR2.cl8) cells, generated in-house at AstraZeneca) were pre-stained with LIVE/DEAD™ Dead Cell Stain (ThermoFisher, Waltham, MA, USA) and incubated with 2 µg/mL of PAR650097 or isotype control antibody (both formatted to human IgG1TM). Cellular binding of PAR650097 or isotype control was detected via direct labelling of antibody with Alexa Fluor 647 Antibody Labeling Kit (ThermoFisher, Waltham, MA, USA) or secondary labelling with Alexa Fluor 488 Anti-Human IgG. Fluorescence was measured on labelled viable cells using a BD LSRFortessa (BD Biosciences, San Jose, CA, usa) cell analyzer/flow cytometer.
Immunocytochemistry
1321N1 parental cells or 1321N1-hPAR2.cl8 cells were grown at 5K cells, per well, in Poly-D-Lysine CELLCOAT 96 Well Cell Culture Microplates (Greiner, Monroe, NA, USA). Cells were labelled with 2 µg/mL PAR650097, or isotype control antibody, for 30 min on ice before washing with PBS, and fixing with formaldehyde for 5 min at room temperature. Cells were washed three times with PBS, before staining with Alexa Fluor 488 Donkey Anti-Human IgG (ThermoFisher, Waltham, MA, USA) at 1:1000 for 30 min on ice. Cells were washed three times with PBS, Hoechst (Life Technologies, Carlsbad, CA, USA) was added to wells in PBS and cells were visualized on an Olympus IX81 Inverted Fluorescense microscope (Olympus, Southend-on-Sea, UK).
Fluorescence imaging plate reader imaging of cytosolic calcium
PAR2 is widely expressed in epithelia and we utilized human lung (A549) and mouse lung (LL/2) epithelial cells (both from ATCC, VA, USA) to characterize the pharmacology of PAR650097. Cells were plated at 5000 (A549) or 7000 (LL/2), per well, in growth medium on PDL-coated Greiner-Bio 384 Well CELLCOAT microplates. Pharmacological responses were measured by detecting PAR-driven (Gq protein-mediated) cellular calcium responses in fluorescence imaging plate reader (FLIPR) assays. Supernatants were replaced with Fluo-4 No Wash Calcium Assay Kit (ThermoFisher, Waltham, MA, USA). Antagonists or agonists were added to dye-loaded cells using the FLIPR Tetra system (Molecular Devices, San Jose, CA, USA) and FLIPR responses (excitation wavelength 470–495 nm/emission wavelength 515–575 nm) were recorded. In antagonistic assay format, when measuring IC50s, cells received antagonist for 30–60 min at room temperature prior to addition of agonist on top. LL/2 cells displayed a very large PAR1 calcium response to thrombin (Figure 1(b)), thus to isolate a trypsin-dependent, PAR2-specific calcium response in LL/2 cells (and enable PAR650097 IC50 generation), thrombin (0.5 nM) was first used to desensitize PAR1. Thrombin (at 0.5 nM) did not affect the subsequent response magnitude to PAR2 stimuli in LL/2 cells (data not shown). Maximum-minimum relative florescence units (RFU) readings from each well were recorded. Concentration-response curves were generated using GraphPad Prism version 7.0 to yield IC50 values. Recombinant human trypsin-1 (Polymun, Klosterneuburg, Austria,), or the PAR2 peptide agonist 2-Furoyl-Leu-Ile-Gly-Arg-Leu-Orn-NH2 (2-furoyl-LIGRLO, Peptides International, Louisville, KY, USA) were used to agonize PAR2. The reverse peptide 2-Furoyl-Orn-Leu-Arg-Gly-Ile-Leu-NH2 (2-furoyl-OLRGIL, Peptides International, Louisville, KY, USA) was used as a negative control. Thrombin from human plasma (Sigma, St. Louis, MO, USA) was used to activate PAR1 where detailed. In some studies, the PAR1 antagonist, SCH79797 (Tocris, Abingdon, UK) or anti-PAR1 inhibitory antibodies, clones ATAP2 (ThermoFisher, Waltham, MA, USA) and WEDE15 (Bechman Coulter, High Wycombe, UK) were used to inhibit PAR1-driven calcium responses.

Fluorescently labelled PAR650097 bound strongly to the human PAR2-expressing 1321N1-hPAR2.cl8 cell line (b), but not to the parental (non-PAR2 expressing) human 1321N1 cells (a), as assessed by flow cytometry and quantified in (c). Immunocytochemistry of the 1321N1-hPAR2.cl8 cell line demonstrated cell surface staining with PAR650097 (d),(e) that was not seen in the parental 1321N1 (no PAR2) cell line (f), or when PAR650097 was replaced with an isotype control in the 1321N1-hPAR2.cl8 cell line (g). Data are from n = 4 ((a)–(c)), or n = 2 ((d)–(g)) independent experiments. Data are presented as mean ± standard error of the mean (SEM) and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (c). * and # represent p < 0.05 in comparison to 1321N1-cl8-hPAR2 cell treated with isotype control antibody and 1321N1 parental cell treated with PAR650097, respectively.
Specificity of PAR650097 for PAR2
The specificity of PAR650097 IgG for human PAR2 over the most closely related PAR family member, PAR1, was determined using a homogeneous time-resolved fluorescence (HTRF™) competition binding assay, in which relative cross-reactivity was assessed by measuring the inhibition of biotinylated PAR2 N-terminal peptide binding to PAR650097 IgG in solution. Binding of biotinylated PAR2 N-terminal peptide to PAR650097 IgG was detected indirectly by time-resolved fluorescence resonance energy transfer (TR-FRET) between europium cryptate labelled streptavidin (CisBio, Bedford, MA, USA), which binds biotinylated PAR2 N-terminal peptide, and an XL665 labelled anti-human-Fc antibody (CisBio, Bedford, MA, USA), which binds PAR650097 IgG. PAR1 and PAR2 were titrated into the assay, and the selectivity of PAR650097 was assessed by measuring the degree of competitive inhibition of biotinylated human PAR2 N-terminal peptide binding to PAR650097 IgG.
Rodent PK
The serum pharmacokinetics of PAR650097 were evaluated in male CD rats following multiple intravenous bolus administrations once weekly for a total of four doses at 10 mg/kg and 100 mg/kg. Serum samples were collected at pre- and post-dose time points for the measurement of PAR650097 concentrations using a Gyrolab method that utilized a sequential flow-through sandwich approach. The serum PAR650097 assay lower limit of quantitation (LLoQ) was 0.0526 µg/mL and any results below the LLOQ were reported as below limit of quantitation (BLQ). Pharmacokinetic parameters of PAR650097 were determined via non-compartmental pharmacokinetic analysis that was performed using Phoenix WinNonlin Professional (version 6.3, Certera Corp., St. Louis, MO). In this study, only serum concentration data following the first PAR650097 intravenous administration is described.
Periorbital and hindpaw frequency response evaluation
Periorbital and hindpaw frequency of response to tactile stimulation were performed prior and after restraint stress (RS), UMB exposure or supradural injections (16). Mice were placed in Plexiglas chambers (4 in L × 4 in W × 8 in H), on top of a wire mesh stand (0.635 cm2 grid), for 2 h. After habituation, 0.4 g and 1 g von Frey filaments (Stoelting, Wood Dale, IL, USA) were applied 10 times to the periorbital and hindpaw region, respectively. The filament was gently applied until the filament slightly arched. Positive responses for the periorbital region were scored after each filament application, as characterized by facial grooming, head shaking, and/or turning away after filament application. Positive responses for the hindpaw region were characterized by sharp withdrawal of the paw, shaking, and/or licking the paw. Further, any such behaviors displayed before filament application were not considered positive responses. The migraine-like pain behavior observed by increased frequency of response was considered to reflect the development of cutaneous allodynia (CA). Frequency response was calculated by [(number of positive responses × 100)/10].
Umbellulone inhalational exposure
Inhalational exposure to UMB was performed as previously described (16). Briefly, a half square gauze (1 in × 1 in) was placed in each nose cone of a multi-station anesthesia board (Parkland Scientific, Coral Springs, FL, USA) and 500 µL of UMB was pipetted onto each gauze. Animals were individually placed at each nose cone under a light anesthesia with 1.5–2% isoflurane. Mice received inhalational exposure to anesthesia for 30 min. Heat pads were placed under the animals during the exposure and animals were observed continuously during the exposure with adjustments of air and isoflurane performed, if needed.
Restraint stress
Mice received episodes of RS for three consecutive days, 2 h each day, using plastic restrainers (Plas-labs INC, 551-BSRR) by pulling the animal’s tail through the restrainer’s stoppers and pushing tightly enough against the animals to limit their movements without causing suffocation. Animals were observed continuously during the stress exposure (16).
Supradural injection
The procedure for supra dura mater injections was performed with slight modification as previously described by Burgos-Vega and colleagues (24), which allows a minimally invasive administration of substances onto the dura mater of mice through the junction of the sagittal and lambdoidal sutures under a light and brief isoflurane anesthesia. Supradural injectors were modified from commercially available cannulas (Plastic One/Invivo1, part #C313I/SPC, Internal Cannula, Standard, 28 gauge). We adjusted the projection length of the injector to 0.7 mm, which is controlled by a stopper to limit the length and maintain dura mater integrity. The injectors were connected to Tygon tubing (Cole Parmer Co. Vernon Hills, Illinois, USA) which was attached to a 25 µL Hamilton syringe (Hamilton, Reno, Nevada, USA). The injector was inserted through the sagittal and lambdoid suture junction region and the injection was delivered onto the dura mater. The total volume injected was 5 µL and each injector was used a maximum of four times.
General experimental design overview
The effect of supradural injection of different doses of SLIGRL in inducing CA was evaluated in naïve female mice. Periorbital and hindpaw frequency of response to tactile stimulation baseline was collected, and SLIGRL or control was injected onto the dura mater. Measurements were made at 5, 10, 20, 40, and 60 min post injection. A different cohort of animals was treated with PAR650097 or control, 72 h prior to supradural injection of SLIGRL. Behavioral measurements were made at 5, 10, 20, 40, and 60 min post SLIGRL injection.
The possible analgesic effect of PAR650097 against migraine-like pain was evaluated using the “two-hit” stress-priming strategy to induce LS to a normally subthreshold UMB stimulus (16). LS was induced with three consecutive episodes of RS in uninjured mice and was revealed by the expression of periorbital and hindpaw CA following inhalational exposure to UMB. Treatment with PAR650097 or isotype control protein was performed 72 h prior to the first RS episode or 72 h prior to the UMB inhalational exposure. Behavioral evaluation was performed on days 3, 5, 7, 9, 11, 14 and 16 after RS. On day 16, animals received inhalational exposure of UMB and measurements were performed 30 min and hourly up to 5 h after the exposure.
In a different cohort of animals, the analgesic effect of PAR650097 or isotype control protein against migraine-like pain was also evaluated using supradural injection of CGRP in naïve mice. The PAR2 mAb or isotype control protein was administered 72 h prior to CGRP injection. Frequency of response to tactile stimulation was collected prior to and after the antibody treatment, and 5, 10, 20, 40, 60, 120 and 180 min after CGRP administration. To evaluate the effect of neutralization of CGRP on CA induced by supradural injection of CGRP and SLIGRL, fremanezumab or vehicle was administered 48 h prior to supradural injections. Baseline to tactile stimulation was collected prior to and after the antibody treatment, and measurements were performed 5, 10, 20, 40, 60, 120 and 180 min or 5, 10, 20, 40 and 60 min after the CGRP or SLIGRL injection, respectively.
Statistical analysis
Statistical analyses used in these studies were calculated using GraphPad Prism 7 (GraphPad Software, La Jolla, CA). Two-way analysis of variance (ANOVA) followed by the Sidak or Tukey test were used for analysis of the time course experiments for two or more sensory threshold groups comparisons, respectively. One-way analysis of variance (ANOVA) followed by Tukey test was used to analyze the flow cytometry quantification and the area over the curve (AOC). The statistical analysis, numbers of animals used (n), p-values and interaction F ratios are reported in Table 1. All data are presented as mean ± standard error of the mean (SEM) and statistical significance was set at p < 0.05.
Summary of statistical analyses.

Results
PAR650097 is a selective, fully inhibitory anti-PAR2 antibody
PAR650097 showed high binding affinity to PAR2 (Supplemental Table 1). Binding of PAR650097 to immobilized human PAR2 demonstrated a KD of 833 pM calculated from on and off rates of 3.8 × 105 M−1 s−1 of k-on and 3.2 × 10−4 s−1 of k-off (Supplemental Table 1). PAR650097 bound strongly to the human PAR2-expressing 1321N1-hPAR2.cl8 cell line (flow cytometry: Figure 1(b) and (c) and Immunocytochemistry: Figure 1((d)–(g)), but not to a control human 1321N1 astrocytoma cell line, lacking PAR2 expression (flow cytometry: Figure 1(a) and (c), and immunocytochemistry: Figure 1(f)). No staining of the human PAR2-expressing 1321N1-hPAR2.cl8 cell line was apparent when an isotype control antibody was substituted for PAR650097 (Figure 1(g)). Human lung epithelial A549 cells (Figure 2(a)), or mouse lung epithelial LL/2 cells (Figure 2(b)) demonstrated robust cellular calcium signaling to both PAR agonistic proteases, trypsin and thrombin, and also to the PAR2 activating peptide agonist, 2f-LIGRLO, but not the control reverse peptide, 2f-OLRGIL. PAR650097 potently inhibited trypsin-driven activation of endogenously expressed PAR2 in human cells (A549; Figure 2(c)), or mouse cells (LL/2; Figure 2(d)) with IC50s of 0.58 or 0.22 nM, respectively.

Human lung epithelial A549 cells (a) or mouse lung epithelial LL/2 cells (b) displayed robust intracellular calcium responses to PAR agonistic proteases (trypsin, thrombin) and to the PAR2 activating peptide (2f-LIGRLO), but not the control reverse peptide (2f-OLRGIL). PAR650097 potently inhibits trypsin-driven activation of endogenously expressed PAR2 in (c) human A549 or (d) mouse LL/2 cells with IC50s of 0.58 or 0.22 nM, respectively. Inhibition curves are presented as mean of two replicates ± SEM and are representative of n = 11 (c) or n = 2 (d) independent experiments.
PAR650097 also revealed high specificity for PAR2 over the most closely related PAR family member, PAR1, demonstrated by lack of effect against thrombin-induced calcium signaling in human A549 cell culture (Supplemental Figure 1(a)). PAR1-specific inhibitory antibodies, ATAP2+WEDE15, or the small molecule PAR1 antagonist, SCH79797, fully blocked thrombin-induced calcium signaling (Supplemental Figure 1(a)). Further specificity of PAR650097 for PAR2 over PAR1 was determined by measuring the degree of inhibition of PAR650097 binding to PAR2, by addition of competing PAR2 or PAR1 N-terminal peptides. PAR650097’s interaction with PAR2 was only out-competed by PAR2 peptide and not PAR1 (at concentrations tested up to 0.8 µM) (Supplemental Figure 1(b)).
PK analysis in rat serum, performed up to 7 days post treatment with PAR650097 (100 mg/kg, i.v.), revealed that following intravenous administration of PAR650097, serum concentrations declined in a biphasic manner with a rapid initial decline, followed by a slower elimination phase. Comparison of PAR650097 levels following a 10 or 100 mg/kg dose, indicated that systemic exposure increased in a dose-proportional manner with linear increases in both Cmax and AUClast observed. Although the half-life of PAR650097 was short (∼1–2 days), circulating concentrations of PAR650097 (when dosed 100 mg/kg, i.v.) remained 964- or 100-fold above in vitro IC50 at 5 and 7 days, respectively (Supplemental Table 2). Whilst efficacy studies were performed in mice and with intraperitoneal dosing, the rat intravenous PK data does show that despite the sub-optimal half-life, systemic exposures equivalent to high multiples of the in vitro IC50 were achievable following in vivo administration of PAR650097. The expectation would be that lower exposure would be observed after intraperitoneal dosing compared with intravenous dosing.
PAR650097, but not isotype control protein, prevents SLIGRL-induced migraine-like pain in mice
Supradural injection of the PAR2 agonist, SLIGRL, induced dose-related CA demonstrated by increased frequency of response after tactile stimulation at the periorbital (Figure 3(a)) and hindpaw (Figure 3(c)) regions. AOC demonstrated the development of dose-related CA induced by SLIGRL (periorbital: Figure 3(b) and hindpaw: Figure 3(d)). Pretreatment with the PAR650097, completely blocked CA produced by SLIGRL (periorbital: Figure 4(a) and hindpaw: Figure 4(c)). The AOC demonstrated the dose-related effect of PAR650097 on the blockade of CA induced by SLIGRL (periorbital: Figure 4(b) and hindpaw: Figure 4(d)). The isotype control protein did not modify CA produced by SLIGRL (Figure 4). These data confirmed specificity in vivo and the anti-allodynic effect of the PAR650097 against the PAR2 agonist.

Supradural injection of PAR2 agonist, SLIGRL, produced dose-dependent CA. (a) Periorbital and (c) hindpaw tactile frequency of response baseline (BL) was collected and animals received supradural injection of different doses of SLIGRL or control (SIF). Measurements were collected on indicated minutes after injection. (b) Periorbital and (d) hindpaw area over the curve (AOC) of the evaluation. Data are presented as mean ± SEM and analyzed using two-way (panels (a) and (c)) and one-way (panels (b) and (d)) ANOVA followed by Tukey’s multiple comparison test (n = 6–17) with * representing p < 0.05 in comparison to the control-treated group.
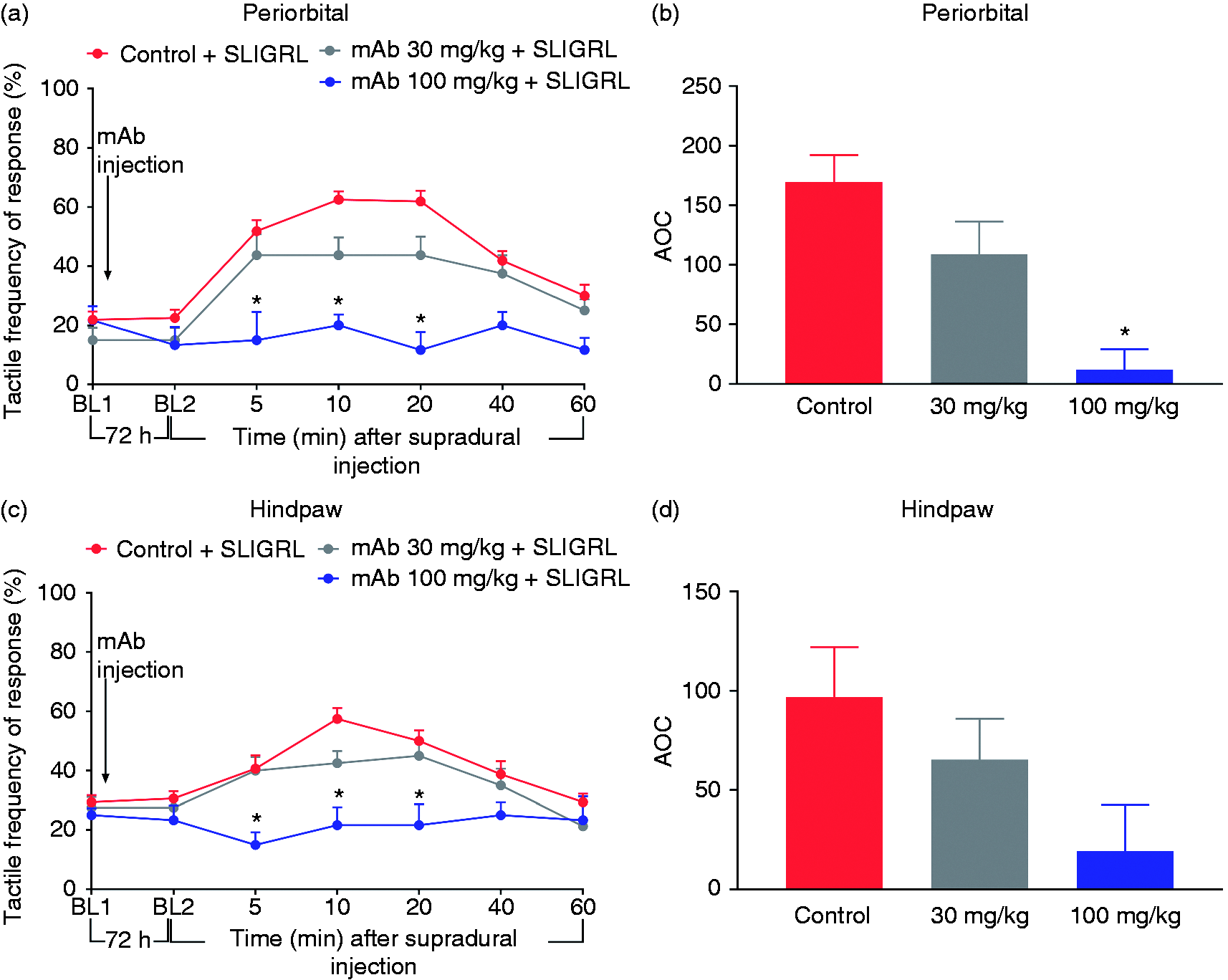
PAR650097 prevented CA induced by supradural injection of SLIGRL. (a) Periorbital and (c) hindpaw tactile frequency of response baseline (BL1) was measured and mice were treated with PAR650097 or isotype control protein. A second baseline (BL2) was assessed after the treatment followed by supradural injection of the highest dose of SLIGRL and measurements performed on indicated minutes after injection. (b) Periorbital and (d) hindpaw area over the curve (AOC) of the evaluation. Data are presented as mean ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (n = 6–16) with * representing p < 0.05 in comparison to control + SLIGRL group.
PAR650097 does not prevent RS priming, but abolishes UMB-induced CA in RS-primed mice
Pretreatment with PAR650097 prior to the RS failed to block either the LS produced by repeated RS, or the CA (periorbital: Figure 5(a) and hindpaw: Figure 5(b)) induced by inhalational exposure to UMB in stress-primed mice. In contrast, pretreatment with PAR650097 prior to UMB exposure completely abolished CA at the periorbital (Figure 5(c)) and hindpaw (Figure 5(d)) regions, induced by the inhalational exposure of UMB in mice primed with RS. The isotype control protein did not modify CA produced by UMB exposure (Figure 5).

Pretreatment with PAR650097 failed to modify stress-priming CA. However, PAR650097 abolished CA when administered immediately before UMB exposure in stress primed mice ((c) and (d)). Periorbital and ((b) and (d)) hindpaw tactile frequency of response baseline (BL, BL1 or BL2) was measured, followed by RS episodes. Behavior was assessed on indicated days post RS. On day 16, baseline (BL) was collected, followed by inhalational exposure to UMB, with measurements collected hourly. PAR650097 or isotype control protein treatments were performed ((a) and (b)) prior to the first RS or ((c) and (d)) prior to inhalational exposure of UMB. Data are presented as mean ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test with * and # representing p < 0.05 in comparison to control + mAb 100 mg/kg versus stress + mAb 100 mg/kg and control + control 100 mg/kg versus stress + control 100 mg/kg groups, respectively (n = 7–12).
PAR650097 abolished migraine-like pain induced by supradural injection of CGRP
Supradural injection of CGRP induced the development of CA (Figure 6). Pretreatment with PAR650097, completely blocked CA induced by CGRP (periorbital: Figure 6(a) and hindpaw: Figure 6(b)). The isotype control protein did not modify CA produced by CGRP (Figure 6).

PAR650097 significantly blocked CA induced by supradural injection of CGRP. (a) Periorbital and (b) hindpaw tactile frequency of response baseline (BL1) was collected and mice were treated with PAR650097 or isotype control protein. A second baseline (BL2) was assessed after the treatment followed by supradural injection of CGRP or control with behavior performed on indicated minutes after injection. Data are presented as mean ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (n = 7–11) with * and # representing p < 0.05 in comparison to mAb 100 mg/kg + control group and control + CGRP group, respectively.
Neutralization of CGRP partially attenuated migraine-like pain induced by supradural activation of PAR2
Periorbital (Figure 7(a)) and hindpaw (Figure 7(b)) CA induced by supradural injection of CGRP was completely blocked by fremanezumab. Pretreatment with this anti-CGRP mAb also appeared to attenuate the CA induced by SLIGRL but the data did not achieve statistical significance (periorbital: Figure 7(c) and hindpaw: Figure 7(d)).

Fremanezumab abolished CA induced by supradural injection of CGRP, but non-significantly attenuated CA produced by SLIGRL. ((a) and (c)) Periorbital and ((b) and (d)) hindpaw tactile frequency of response baseline (BL1) was collected and mice were treated with fremanezumab or control. A second baseline (BL2) was assessed after the treatment followed by supradural injection of ((a) and (b)) CGRP or ((c) and (d)) SLIGRL, followed by behavior evaluation on indicated minutes after injection. Data are presented as mean ± SEM and analyzed using two-way ANOVA followed by Tukey’s multiple comparison test (n = 8–12) with * representing p < 0.05 in comparison to control + CGRP group.
Discussion
We have characterized a humanized mAb with full functional activity and high selectivity at PAR2, and attempted to identify the potential contribution of this receptor in pain and sensitization related to migraine. Several findings are notable from our studies. First, blockade of PAR2 completely blocked periorbital and hindpaw CA elicited by supradural CGRP. This observation suggests that activation of the PAR2 receptor is a downstream consequence of exogenous, and possibly endogenous, CGRP. Second, consistent with this conclusion, CGRP sequestration with fremanezumab at a dose that fully prevented the effects of supradural CGRP only partially modified CA induced by the PAR2 agonist, SLIGRL, suggesting that migraine-like pain may be initiated both by CGRP-dependent and -independent mechanisms. Third, we found that while PAR2 blockade did not prevent the development of LS induced by stress priming, it was able to prevent CA induced by TRPA1 activation in animals with LS. Collectively, these findings suggest that PAR2 blockade is a promising strategy for preventive migraine therapy.
PAR650097 is a novel mAb that fully antagonizes PAR2 function in human and rodent cells. The IC50 of 0.58 (human) or 0.22 nM (mouse) of PAR650097 in inhibiting trypsin-driven activation of endogenously expressed PAR2 in human and mouse cells confirms high affinity for the receptor. The specificity of PAR650097 is supported by the lack of significant activity at the PAR1 receptor. The current data established PAR650097 as a novel mAb with high selectivity for PAR2 and a PK profile that, if further optimised, may be progressed to human studies.
Preclinical studies have previously demonstrated the participation of PAR2 in initiation and/or maintenance of pain, as well as in headache (13,14,25–28). Herein, we explored the potential contribution of PAR2 in the development of LS and expression of CA relevant to migraine-like pain. Supradural administration of the PAR2 agonist SLIGRL elicited CA, which was blocked by PAR650097, confirming in vivo selectivity. These findings are consistent with previous reports that demonstrated migraine-like pain following supradural injection of a highly selective PAR2 agonist, and the complete ablation of these phenotypes in transgenic mice lacking PAR2 expression, as well as by a novel small molecule PAR2 antagonist or sumatriptan (13).
Stress, or relief of stress, is considered as an important trigger of a migraine attack (29,30). The frequency of migraine attacks is powerful risk factor for migraine chronification, suggestive of a priming effect in people with migraine (31–33). In our previous study, repeated stress promoted a state of LS in which CA is elicited by a subthreshold challenge with the environmental irritant UMB (16), a TRPA1 agonist known to induce headache attacks in individuals with underlying headache disorders (34). We found that blockade of PAR2 with PAR650097, prior to the first RS, did not prevent CA during the stress priming sessions. However, PAR2 blockade prevented UMB-induced CA in animals with LS when administered immediately before UMB exposure. These data suggest that blockade of PAR2 is downstream of the pain-inducing consequences of activation of TRPA1 in the trigeminal system. It may be speculated that in animals with LS, UMB leads to dural release of pain-promoting substances, including neuropeptides such as CGRP, as well as degranulation of mast cells and consequent activation and/or sensitization of PAR2 (14,25–27,35). PAR2 activation has been reported to contribute to activation of trigeminal afferents by promoting release of CGRP and/or mast cell degranulation (14,15,27).
As a result of recent advances in the understanding of migraine pathophysiology, anti-CGRP peptide and CGRP receptor mAbs, along with small molecule CGRP receptor antagonists, are currently approved and in use as preventive or acute anti-migraine strategies (36–38). Preclinically, direct application of CGRP onto the dura mater of female mice produces migraine-like phenotypes such as CA, ongoing pain and priming to subsequent migraine triggers (17). Our previous study demonstrated that the CGRP receptor antagonist, olcegepant, was not effective in reversing established CA induced by UMB challenge in animals with LS induced by stress. However, pre-treatment with olcegepant before inducing pain with UMB prevented the expression of CA, suggesting that the CGRP receptor might be a pivotal contributor to the initiation of migraine-like pain, but is only one factor that is involved in the maintenance of pain (16). Multiple lines of data support the evidence that CGRP plays an important, but not exclusive, role in migraine headache (39). The present study demonstrated that supradural injection of CGRP produced CA in female mice that was fully blocked by fremenezumab, an anti-CGRP mAb. Interestingly, pretreatment with PAR650097 also fully blocked CA induced by CGRP. These data support the importance of PAR2 in this preclinical model of migraine and suggest that PAR2 activation likely plays a significant role in the CGRP-mediated initiation of migraine-like pain. In contrast, pre-treatment with fremanezumab at a dose that fully blocked the allodynic actions of supradural CGRP only partially attenuated CA induced by supradural SLIGRL, without achieving statistical significance. These data support the likelihood of non-CGRP dependent mechanisms that can initiate migraine-like pain behaviors. Consistent with this hypothesis, Bhatt and colleagues have shown that PAR2 activation leads to vasodilation of dural arteries in rats and these responses are partially mediated by nitric oxide, but are independent of CGRP and mast cell-mediated mechanisms (40). Collectively, these findings support the contributions of CGRP-dependent and independent processes to migraine pain pathophysiology.
Limitations of our study include the use of SLIGRL, which is not completely selective to PAR2 (41). SLIGRL is also a Mas-related GPCR agonist and the Mrg family of receptors expressed in sensory neurons and mast cells (42). While modulation of these receptors may also contribute to the migraine-like pain, we note that PAR650097 is highly selective for PAR2 receptors, which diminishes concerns regarding additional non-PAR2 mechanisms of SLIGRL in vivo. Additionally, our study only examined female mice and it is not known if similar results would be observed in male animals. Improvements of the half-life of PAR2 antibodies should also be addressed in future studies. Finally, the pharmacokinetic determinations were made in rats following intravenous administration, but the in vivo behavior was carried out in mice with intraperitoneal administration. Differences in pharmacokinetics in the two species and with these two routes of administration are to be expected. Nevertheless, the pretreatment times chosen showed specificity for inhibition of PAR2 activation consistent with the in vitro characterization of PAR650097.
Results from clinical studies demonstrate the anti-CGRP mAbs (peptide or receptor) and the small molecule antagonists significantly reduce migraine headache days per month in about 50% of people with migraine (37,43–45). However, these treatments do not completely abolish migraine attacks in responders, and for a considerable percentage of people these therapies are not effective (37,38,43). The outcomes of these studies suggest that a substantial number of individuals with migraine are non-responders to the anti-CGRP therapies, additionally supporting the contribution of CGRP-independent processes involved in migraine pain pathophysiology (43). Data from the current investigation suggest that a PAR2 mAb could represent a preventive therapeutic strategy that could be effective against both CGRP-dependent and independent migraine-initiation mechanisms. PAR2 blockade might represent a novel pharmacological strategy in migraine prevention.
Article highlights
Discovery and in vitro characterization of a selective fully humanized PAR2 mAb. PAR2 activation results from exogenous CGRP, suggesting a downstream mechanism. PAR2 activation can also promote migraine-like pain independently of CGRP. PAR2 mAb may represent a novel, non-CGRP preventive strategy to control migraine-like pain.
Supplemental Material
sj-pdf-1-cep-10.1177_0333102420966581 - Supplemental material for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine
Supplemental material, sj-pdf-1-cep-10.1177_0333102420966581 for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine by Caroline M Kopruszinski, Peter Thornton, Joanne Arnold, Philip Newton, David Lowne, Edita Navratilova, Juliana Swiokla, David W Dodick, Claire Dobson, Ian Gurrell, Iain Patrick Chessell and Frank Porreca in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_0333102420966581 - Supplemental material for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine
Supplemental material, sj-pdf-2-cep-10.1177_0333102420966581 for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine by Caroline M Kopruszinski, Peter Thornton, Joanne Arnold, Philip Newton, David Lowne, Edita Navratilova, Juliana Swiokla, David W Dodick, Claire Dobson, Ian Gurrell, Iain Patrick Chessell and Frank Porreca in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_0333102420966581 - Supplemental material for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine
Supplemental material, sj-pdf-3-cep-10.1177_0333102420966581 for Characterization and preclinical evaluation of a protease activated receptor 2 (PAR2) monoclonal antibody as a preventive therapy for migraine by Caroline M Kopruszinski, Peter Thornton, Joanne Arnold, Philip Newton, David Lowne, Edita Navratilova, Juliana Swiokla, David W Dodick, Claire Dobson, Ian Gurrell, Iain Patrick Chessell and Frank Porreca in Cephalalgia
Footnotes
Acknowledgements
The authors wish to thank Nils-Olov Hermansson, AstraZeneca for generating the 1321N1 cells overexpressing human PAR2. David Lowne’s current address is Immunocore Limited, 101 Park Dr, Milton, Abingdon OX14 4RY, UK.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PT, JB, PN, DL, CD, IG and IC are employees of AstraZeneca. CK received postdoctoral support from AstraZeneca. FP received laboratory support for this study from AstraZeneca. DWD reports the following conflicts within the past 12 months: Consulting: AEON, Amgen, Clexio, Cerecin, Allergan, Alder, Biohaven, Linpharma, Lundbeck, Promius, Eli Lilly, eNeura, Novartis, Impel, Theranica, WL Gore, Nocira, XoC, Zosano, Upjohn (Division of Pfizer), Pieris, Revance, Equinox. Honoraria: CME Outfitters, Curry Rockefeller Group, DeepBench, Global Access Meetings, KLJ Associates, Majallin LLC, Medlogix Communications, Miller Medical Communications, Southern Headache Society (MAHEC), WebMD Health/Medscape, Wolters Kluwer, Oxford University Press, Cambridge University Press. Research Support: Department of Defense, National Institutes of Health, Henry Jackson Foundation, Sperling Foundation, American Migraine Foundation, Patient Centered Outcomes Research Institute (PCORI). Stock options/shareholder/patents/board of directors: Aural Analytics (options), ExSano (options), Palion (options), Healint (options), Theranica (options), Second Opinion/Mobile Health (options), Epien (options/board), Nocira (options), Ontologics (options/board), King-Devick Technologies (options/board), Precon Health (options/board). Patent 17189376.1-1466:vTitle: Botulinum Toxin Dosage Regimen for Chronic Migraine Prophylaxis.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by an unrestricted grant from AstraZeneca to FP.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.