
Abstract
Introduction
Based on the abundant preclinical and human evidence for the key role of calcitonin gene related peptide (CGRP) in the neurobiology and underlying mechanism of migraine, small molecule antagonists at the CGRP receptor were developed and all were shown to be effective for the acute treatment of migraine. In fact, several CGRP receptor antagonists have been demonstrated in phase II studies to be effective for the acute treatment of migraine (1–5). Recently, pivotal phase III studies evaluating the efficacy and safety of rimegepant and ubrogepant have confirmed earlier phase studies with these molecules, demonstrating efficacy and a favorable tolerability profile (6–7).
Key differences between small molecule drugs and monoclonal antibodies.

Attributes of emerging a-CGRP monoclonal antibodies.

Efficacy, safety and tolerability of CGRP mAbs
Erenumab
Erenumab (formerly AMG 334) is a fully human IgG2 mAb that selectively targets the canonical CGRP receptor. Its safety, tolerability and efficacy have been evaluated in several phase 2 and 3 controlled clinical trials. In three different monthly subcutaneous doses (7 mg, 21 mg, or 70 mg), erenumab was evaluated for the preventive treatment of episodic migraine in a multicenter, randomized, double-blind, placebo-controlled, phase 2 trial that included 483 adult patients with 4 to 14 migraine days per month. Patients were randomly assigned in a 3:2:2:2 ratio (NCT01952574) (9). The mean change in monthly migraine days (MMDs) at weeks 9–12 (primary endpoint), was −3.4 (SE 0.4) days with 70 mg versus −2.3 (0.3) days with placebo (difference −1.1 days [95% CI –2.1 to −0.2], p = 0.021). The 7 mg and 21 mg doses were not significantly different than placebo. The 50% responder rate (the proportion of patients with > 50% reduction in MMDs) for the 70 mg dose group was significantly greater than placebo at week 4 (38% vs. 23%; OR 2.1 [95% CI 1.2–3.7], p = 0.008), week 8 (46% vs. 33%; OR 1.7 [95% CI 1.0–2.8], p = 0.058) and at the primary endpoint, week 12 (46% vs. 30%; OR 2.0 [95% CI 1.2–3.4], p = 0.011. Adverse events were recorded in 54% of patients who received placebo, 50% of patients in the 7 mg group, 51% of patients in the 21 mg group, and 54% of patients in the 70 mg group. The most frequently reported adverse events were nasopharyngitis, fatigue, and headache. No treatment-related serious adverse events were reported, there were no clinically significant abnormalities in vital signs, laboratory parameters, or electrocardiograms, and the percentage of patients who withdrew from the study because of adverse events in the placebo, 7 mg, 21 mg, and 70 mg groups respectively were low at 1%, 2%, 2%, and 3%.
The efficacy and safety of erenumab for the preventive treatment of chronic migraine (CM) was evaluated in a phase 2 randomized, double-blind, placebo-controlled, multicenter trial. Six hundred and sixty seven adults aged 18–65 years were randomly assigned (3:2:2) to subcutaneous placebo, erenumab 70 mg or 140 mg, administered monthly for 12 weeks (NCT02066415) (10). Patients reported mean MMDs of 18.2, 17.9, and 17.8 at baseline in the placebo, erenumab 70 mg, and erenumab 140 mg groups, respectively. Criteria for medication overuse were met in 41% of the patients in this trial. Significant reductions in MMDs versus placebo were demonstrated in both dose groups (−6.6 days vs. placebo −4.2 days; difference −2.4 [95% CI −3.5 to −1.4], p < 0.0001). The responder rates (70 mg [40%], 140 mg [41%], placebo [23%]) and odds of achieving a ≥50% reduction in MMDs during the last month of the 12-week trial were significantly greater for both erenumab doses compared with placebo (70 mg OR 2.2 [95% CI 1.5–3.3], p = 0.0001; 140 mg OR 2.3 [95% CI 1.6–3.5], p < 0.0001). The frequency of adverse events in the placebo (39%), 70 mg (44%), and 140 mg (47%) were similar; the most frequent adverse events being injection-site pain, upper respiratory tract infection, and nausea. No clinically significant abnormalities in vital signs, laboratory results, or electrocardiograms were found and only four patients (two in the placebo and two in the 140 mg dose group) withdrew because of adverse events. No patient developed neutralizing antibodies, while 11 patients in the 70 mg group and three patients in the 140 mg group had anti-erenumab binding antibodies. There was no relationship between the presence of binding antibodies and adverse events or efficacy.
The preliminary findings in episodic migraine were confirmed in two recently completed randomized, double-blind, placebo-controlled, phase 3 studies. In the first (ARISE: NCT02483585), 577 adult patients were randomized to monthly subcutaneous injections of placebo or 70 mg erenumab for 3 months (11). A significant reduction in monthly migraine days during weeks 9–12 in the erenumab group (−2.9) compared to placebo (−1.8) was demonstrated (LSM (95% CI) treatment difference of 1.0 (1.6, 0.5) (p < 0.001). In addition, significantly more patients who received erenumab 70 mg experienced a ≥ 50% reduction in MMDs compared to placebo (39.7% and 29.5%, (OR: 1.59 (95% CI: 1.12, 2.27) (p = 0.010). The most common adverse events were upper respiratory tract infection, injection site pain, influenza, fatigue, nausea, migraine, sinusitis, nasopharyngitis, and constipation (reported in 2% of patients in either group). No significant changes in vital signs, laboratory values, or electrocardiograms were identified and adverse events that led to study withdrawal occurred in one (0.3%) patient in the placebo group and five (1.8%) patients in the erenumab 70 mg group. Anti-erenumab binding antibodies at week 12 occurred in 12 (4.3%) patients and one patient developed neutralizing antibodies at week 4, but only transiently.
In the second phase 3 episodic migraine trial (STRIVE), 955 patients were randomized to a subcutaneous injection of either placebo, erenumab 70 mg or erenumab 140 mg for 6 months (NCT02456740) (12). The MMDs at months 4–6 was significantly reduced in both the 70 mg (−3.2) and 140 mg (−3.7) dose groups compared to placebo (−1.8; p < 0.001 for each dose vs. placebo). A significantly higher percentage of patients experienced ≥ 50% reduction in MMDs during months 4–6 in the 70 mg (43.3%) and 140 mg (50%) erenumab dose groups, compared to placebo (26.6%; p < 0.001 for each dose vs. placebo). All other secondary endpoints in this trial were met including a significant reduction in the number of days of use of acute migraine-specific medication, physical impairment and everyday activities scores. As in previous studies, the frequency and severity of adverse events, serious adverse events, and adverse events that resulted in withdrawal from the study were similar between the 70 mg and 140 mg erenumab (2.2%, 2.2%) and placebo (2.5%) groups.
In a long-term (5-year) open-label extension study designed to assess long-term safety and efficacy, a one-year pre-planned interim analysis was reported in 383 patients who had a median exposure to erenumab 70 mg of 575 days (range 28–822 days) (13). The mean MMDs at baseline in the parent study was 8.2 (2.6) days, 6.3 (4.2) at the end of the double-blind phase at 12 weeks, and 3.7 (4.0) at week 64. At week 64, ≥ 50%, ≥ 75%, and 100% reductions in mean MMDs were achieved by 65%, 42%, and 26% of patients, respectively. Mean disability (HIT-6) scores declined from a mean of 60.2 (6.3) at baseline and 51.7 (9.2) at week 64. The tolerability and safety profile of erenumab remained consistent with that seen in the erenumab and placebo groups during the double-blind phase. Development of neutralizing antibodies (NABs) against erenumab occurred in nine (2.4%) patients on at least one occasion, but there was no association between the presence of NABs and either efficacy or safety findings.
An important consideration in the use of CGRP mAbs will be the extent to which they are effective in patients who have failed to respond to prior preventive drugs. In the STRIVE trial, a subgroup analysis of patients who had previously failed one (369) or two (161) prior oral preventive drugs due to lack of efficacy or intolerability was conducted (14). The odds of achieving a ≥ 50% reduction in mean MMDs between 4–6 months after starting therapy was 2.9 times higher for the 70 mg dose group for both treatment failure groups and 3.1 and 4.5 times higher for the 140 mg dose group in those failing one or two prior preventives, respectively. These preliminary data were confirmed in the recently completed LIBERTY trial (NCT03096834). LIBERTY was a phase 3b multicenter, randomized 12-week, double-blind, placebo-controlled study evaluating the safety and efficacy of erenumab in patients with episodic migraine who failed two to four prior oral preventive drugs. In this study, 246 patients were randomized to erenumab 140 mg or placebo. The proportion of patients at baseline who failed two, three, and four previous treatments were 38.6%, 37.8%, and 22.8% respectively. The proportion of patients who experienced a 50% reduction in MMDs was 30.3% for the 140 mg dose group compared to 13.7% for placebo (OR 2.73 [95% CI 1.43–5.19]; p = 0.002). Safety and tolerability were comparable to placebo, and no patients in the erenumab cohort discontinued due to adverse events.
Eptinezumab
Eptinezumab (formerly ALD 403) is a genetically engineered, desialylated, humanized a-CGRP IgG1 antibody that selectively and potently binds to α and β forms of human CGRP. In a randomized double-blind phase 2 trial in 174 adult patients with episodic migraine (NCT01772524), patients received, in a 1:1 ratio, either 1000 mg eptinezumab or placebo intravenously (15). The mean change in migraine days between baseline and weeks 5–8 (primary efficacy endpoint) was −5.6 (SD 3.0) for the eptinezumab group compared with −4.6 (3.6) for the placebo group (difference −1.0 [95% CI −2.0 to 0.1]; one-sided p = 0.0306). The mean change from baseline in migraine days per month across weeks 1–12 was −5.5 (SD 3.1) for eptinezumab and −4.4 (3.1) for placebo. The ≥ 50%, ≥ 75% and 100% responder rates over the 12-week trial period were 61%, 33%, and 16% vs. 33%, 9%, and 0% for placebo. Similar rates of adverse events were experienced in the placebo group (52%) and eptinezumab group (57%) with the most common adverse events being upper respiratory tract infection, urinary tract infection, fatigue, back pain, nausea and vomiting, and arthralgia. There were no serious adverse events attributable to the study drug. There were no infusion reactions, and no significant differences in vital signs, 12-lead ECGs, or laboratory parameters.
In a randomized, double-blind, placebo-controlled, phase 2b dose-ranging study (NCT02275117), 616 patients with chronic migraine were randomized in a 1:1:1:1:1 ratio to eptinezumab 300 mg, 100 mg, 30 mg, 10 mg, or placebo administered as a single intravenous infusion (16). The primary endpoint ( ≥ 75% decrease in MMDs over weeks 1–12) for eptinezumab 300 mg, 100 mg, 30 mg, and 10 mg were 33.3%, 31.4%, 28.2%, and 26.8% respectively, vs. 20.7% for placebo (p = 0.033; 0.0715; 0.2013; 0.2938 vs. placebo). The primary endpoint results of this study showed that only the 300 mg dose was superior to placebo while the more conventional 50% responder rates demonstrate that lower doses may be effective. The reason(s) for the lack of efficacy of lower doses is unclear, but the very high placebo response rate for such a robust endpoint is unusual. The ≥ 50% responder rates over weeks 1–12 were 57.0%, 55.1%, 55.6% and 43.9% in the eptinezumab 300 mg, 100 mg, 30 mg, and 10 mg groups, compared with 40.5% in the placebo group (p = 0.013, 0.034, 0.027, 0.55). The mean number of MMDs was reduced from 16.5 at baseline to 8.3 over weeks 1–12 in the eptinezumab 300 mg group compared to a reduction from 16.4 to 10.9 days in the placebo group (p = 0.003). The incidence of adverse events was similar in active and placebo groups respectively, and there were no serious adverse events related to the study drug. Ten subjects had treatment-emergent adverse events leading to interruption of eptinezumab infusion, including six subjects with hypersensitivity. Hypersensitivity symptoms included sneezing, cough, nasal congestion, scratchy throat, burning and watery eyes, edema in the face and eyelids, shortness of breath, hives, and itching. Each of these symptoms were mild to moderate in severity and were treated successfully with an antihistamine or corticosteroid within 24 hours.. There were no significant changes in laboratory tests, vital signs, 12-lead ECGs, or physical examination findings.
Eptinezumab was also evaluated in two pivotal phase 3 trials in episodic (PROMISE 1; NCT02559895;
According to the press release for PROMISE 2 (
Fremanezumab
Fremanezumab (formerly TEV-48125, LBR-101 and RN-307) is a fully humanized IgG2a monoclonal antibody that potently and selectively binds to the CGRP ligand. In a large, randomized, double-blind, double-dummy, placebo-controlled, parallel-group phase 2b study (NCT02021773), 264 adult patients with chronic migraine were randomly assigned (1:1:1) to receive monthly subcutaneous injections of fremanezumab 675/225 mg (675 mg in the first treatment cycle and 225 mg in the second and third treatment cycles), 900 mg (900 mg in all three treatment cycles), or placebo (18). The primary endpoint (mean change from baseline in number of headache-hours during weeks 9–12) was –59.84 hours (SD 80.38) in the 675/225mg group and –67.51 hours (79.37) in the 900mg group, compared with −37.10 hours (79.44) in the placebo group. The least square mean difference in the reduction of headache-hours between the placebo and 675/225 mg dose groups was −22.74 hours (95% CI −44.28 to −1.21; p = 0.0386), whereas the difference between placebo and 900 mg dose groups was −30.41 hours (−51.88 to −8.95; p = 0.0057). The least square mean (LSM) change in the number of moderate or severe headache-days from baseline was a secondary endpoint in this trial and after the third treatment cycle the LSM change was −4.2 days (SD 6.32) in the placebo group, −6.04 days (6.41) in the 675/225 mg group, and −6.16 days (6.32) in the 900 mg group. Treatment-emergent adverse events (TEAEs) were reported by 40% of patients receiving placebo, 53% of patients receiving 675/225 mg, and 48% of patients receiving 900 mg. The most common adverse events were mild injection-site reaction and pruritus. No significant changes in vital signs or laboratory tests were reported and no serious adverse events occurred that were considered to be treatment related. Treatment-related adverse events, most of which were injection site reactions, occurred in 17% of patients in the placebo group, 29% of patients in the 675/225 mg group, and 32% of patients in the 900 mg group.
In a post-hoc analysis of this study, the 675/225 mg dose was superior to placebo as early as day 7. The 900 mg dose separated from placebo 3 days after treatment was administered (p = 0.048 and p = 0.033, respectively) (19). In addition, the improvement was sustained through the second (p = 0.004 and p < 0.001) and third (p = 0.025 and p < 0.001) weeks and throughout the study (month 3, p = 0.0386 and p = 0.0057). Also, in a post-hoc analysis, a greater percentage of patients (53%) in the 675/225 mg group and 900 mg group (55%) had more than a 50% improvement in the number of days with moderate to severe headaches in weeks 9–12 as compared to the placebo group (31%; p < 0.001) (18).
In a multicenter, randomized, double-blind, placebo-controlled, phase 2b study (NCT02025556), fremanezumab was evaluated in patients with high-frequency episodic migraine (HFEM; 8–14 migraine headache days per month). Two hundred and ninety seven adult patients were randomly assigned in a 1:1:1 ratio to receive monthly subcutaneous injections of fremanezumab 225 mg, 675 mg, or placebo (20). The LSM change in the number of migraine days from baseline to weeks 9–12 was −3.46 days (SD 5.40) in the placebo group, −6.27 days (5.38) in the 225 mg dose group, and −6.09 days (5.22) in the 675 mg dose group. The LSM difference in migraine-day reduction between placebo and 225 mg dose groups was −2.81 days (95% CI −4.07 to −1.55; p < 0.0001), while the difference between the placebo and 675 mg dose group was −2.64 days (−3.90 to −1.38; p < 0.0001). Responder rates were assessed in a post-hoc analysis. The proportion of patients with ≥ 50% reduction in the number of migraine days was 28% in the placebo group, 53% in the 225 mg group (p = 0.0005) and 59% of patients in the 675 mg group (p < 0.0001). The proportion of patients with ≥ 75% or greater reduction in the number of migraine days was 11% in the placebo group, 34% in the 225 mg group (p = 0.0001) and 31% in the 675 mg group (p = 0.0008). In this trial, preventive drug use was present in 27%, 34%, and 27% in the placebo, 225 mg, and 675 mg dose groups respectively. The proportion of patients who had previously discontinued preventive drug use because of poor efficacy or tolerability was 27%, 33% and 25% in the placebo, 225 mg, and 675 mg dose groups respectively. In the subset of patients who were not taking migraine preventive drugs, the 50% responder rates were 22%, 66% (p = 0.0004) and 67% (p = 0.0002) in the placebo, 225 mg, and 675 mg dose groups, while the 75% responder rates were 8%, 48%, and 36% in the 225 mg group (p = 0.0003) and 675 mg group (p = 0.0048) dose groups. Adverse events occurred in 58 (56%) patients in the placebo group, 44 (46%) patients in the 225 mg dose group, and 57 (59%) patients in the 675 mg dose group. Moderate or severe adverse events were reported for 29 (27%) patients, 24 (25%) patients, and 26 (27%) patients. Treatment-emergent adverse events were similar across all groups and reported by 56%, 46% and 59% of patients in the placebo, 225 mg and 675 mg groups. Treatment-related adverse events were also very similar across all groups and were reported by 23%, 27%, and 25% of patients in the placebo, 225 mg, and 675 mg groups. There were no treatment-related serious adverse events and no significant changes in laboratory tests, vital signs, blood pressure, or 12-lead ECGs among groups. Two (1%) patients tested positive for antibodies against fremenezumab before and after taking the study drug, with no rise in antibody titers.
Fremanezumab was evaluated for the preventive treatment of episodic and chronic migraine in two pivotal phase 3 trials (HALO). In the HALO chronic migraine trial (NCT02621931), 1130 adult patients were randomized in a 1:1:1 ratio to receive fremanezumab quarterly (a single dose of 675 mg at baseline and placebo at weeks 4 and 8), fremanezumab monthly (675 mg at baseline and 225 mg at weeks 4 and 8), or matching placebo by subcutaneous administration (21). At baseline, the mean number of headache days (defined as days in which headache pain lasted ≥ four consecutive hours and had a peak severity of at least a moderate level or days in which acute migraine–specific medication (triptans or ergots) was used to treat a headache of any severity or duration) per month was 13.2, 12.8, and 13.3 respectively. The least-squares mean ( ± SE) reduction in the average number of headache days per month was 4.3 ± 0.3 with fremanezumab quarterly, 4.6 ± 0.3 with fremanezumab monthly, and 2.5 ± 0.3 with placebo (p < 0.001 for both comparisons with placebo). The percentage of patients with a reduction of at least 50% in the average number of headache days per month was 38% in the fremanezumab-quarterly group, 41% in the fremanezumab-monthly group, and 18% in the placebo group (p < 0.001 for both comparisons with placebo). Adverse events were reported for 64% of the patients receiving placebo, 70% of those receiving fremanezumab quarterly (p = 0.06 vs. placebo), and 71% of those receiving fremanezumab monthly (p = 0.03 vs. placebo). Adverse events were mild in over 90%, and study discontinuation due to adverse events occurred in 1% in the fremanezumab-quarterly group, 2% in the fremanezumab-monthly group, and 2% in the placebo group. Injection-site reactions were reported in 40% of the patients receiving placebo, 47% of those receiving fremanezumab quarterly (p = 0.08 vs. placebo), and 47% of those receiving fremanezumab monthly (p = 0.03 vs. placebo). The severity of injection-site reactions did not differ significantly among the trial groups. No participants had anaphylaxis or a severe hypersensitivity reaction. Antidrug antibodies developed in two patients who received fremanezumab quarterly. No clinically significant changes in vital signs, physical-examination findings, or 12-lead ECG results occurred in any of the trial groups.
In a randomized, double-blind, placebo-controlled, parallel-group phase 3 trial, the efficacy and safety of fremaneuzumab for the preventive treatment of episodic migraine (EM) was conducted in patients with 6–14 headache days (and at least four migraine days) per month confirmed during the 28-day pre-treatment period (HALO EM; NCT02629861) (22). A total of 875 patients were randomized 1:1:1 to receive subcutaneous monthly dosing of fremanezumab (225 mg at baseline, weeks 4 and 8), a single higher dose of fremanezumab, intended to support a quarterly dose regimen (675 mg at baseline, placebo at weeks 4 and 8), or placebo (at baseline, weeks 4 and 8). The MMDs were reduced from 8.9 days to 4.6 days in the fremanezumab monthly dosing group, from 9.2 to 4.9 days in the fremanezumab single-higher-dose group, and from 9.1 days to 5.9 days in the placebo group. This resulted in a difference for monthly dosing vs. placebo of −1.5 days (95% CI, −2.01, −0.93; p < 0.001) and with single higher dosing vs. placebo of −1.3 days, (95% CI, −1.79, −0.72; p < 0.001). The proportion of patients with ≥ 50% reduction in MMDs during the 12-week treatment period were 47.7% for the fremanezumab monthly dosing group (difference vs. placebo: 19.8% [95% CI: 12.0, 27.6], p < 0.001) and 44.4% for the fremanezumab single-higher-dose group (difference vs. placebo: 16.5% [95% CI: 8.9, 24.1], p < 0.001) compared with 27.9% for the placebo group. Treatment-related adverse events occurred in 48%, 47%, and 37% in the fremanezumab monthly, quarterly, and placebo groups respectively. The most common adverse events in patients treated with fremanezumab were injection-site reactions with injection-site pain, induration, and erythema occurring in 25.9%, 15.4%, and 14.0% of the fremanezumab monthly, quarterly, and placebo groups respectively. The proportion of patients who discontinued due to adverse events was similar in each treatment group (2%). No significant changes in laboratory parameters, vital signs, physical examination findings, or 12-lead ECG changes were seen. Anti-drug antibodies were found in four patients in the fremanezumab monthly dosing group, but this was not associated with any adverse events.
In a post-hoc analysis of the two phase 2 placebo-controlled studies in EM and CM (NCT020255556 and NCT02021772), the efficacy of fremanezumab as an add-on therapy in patients already taking oral preventive drugs was evaluated (23). In a sample of 133 patients, (67 fremanezumab and 66 placebo), the reduction in migraine days was 12.4 for fremanezumab and 7.4 for placebo (p = 0.0321). Decreases in moderate/severe headache days (12.5 vs. 7.1, p = 0.0058), was also demonstrated. Treatment-emergent adverse events were generally mild and transient, and no serious adverse events were considered to be treatment-related. Although very preliminary, this analysis suggests that fremanezumab may be effective as an add-on treatment for migraine patients who are already being treated with other oral migraine preventive drugs.
Galcanezumab
Galcanezumab (formerly LY2951742) is a fully humanized anti-CGRP IgG4 mAb that targets the CGRP ligand and binds with high affinity (KD = 31 pM) and high specificity ( > 10,000-fold vs. related peptides adrenomedullin, amylin, calcitonin and intermedin).
A randomized, double-blind, placebo-controlled, phase 2 proof-of-concept trial (NCT01337596) was conducted in 281 adult patients with episodic migraine (4–14 migraine headache days per month) (24). Patients were randomly assigned (1:1) to galcanezumab 150 mg or placebo by subcutaneous injection once every 2 weeks for 12 weeks. The mean change from baseline to week 12 in the number of migraine headache days was −4.2 (SD 3.1; 62.5% decrease) in the LY2951742 group compared with −3.0 (SD 3.0; 42.3% decrease) in the placebo group (LSM difference −1.2 [90% CI −1.9 to −0.6], p = 0.0030). The proportion of patients with ≥50% reduction in MHDs was greater in the galcanezumab group (70%) compared to the placebo group (45%; OR 2.88 [90% CI 1.78–4.69]). In an exploratory post-hoc analysis, 49% of galcanezumab-treated patients experienced a ≥ 75% reduction in MHDs compared to 27% in the placebo group (OR 2.54, 90% CI 1.56–4.13) while 32% in the galcanezumab group vs. 17% in the placebo group achieved a 100% reduction in MHDs (OR 2.16 [90% CI 1.24–3.75]). Adverse events that occurred more frequently with galcanezumab than with placebo included injection site pain, erythema, or both (21 [20%] of 107 vs. seven [6%] of 110), upper respiratory tract infections (18 [17%] vs. 10 [9%]), and abdominal pain (six [6%] vs. three [3%]). There were no serious adverse events that were deemed related to the study drug, and no patients in the galcanezumab group withdrew because of an adverse event. There were no clinically important changes in laboratory parameters, ECGs, or vital signs between the groups. Anti-drug antibodies were detected in eight patients at screening, and 20 patients at the end of the study.
In another phase 2b dose-ranging study, galcanezumab 5 mg, 50 mg, 120 mg, 300 mg, or placebo were subcutaneously administered monthly over a 3-month treatment period in 936 adult patients with episodic migraine (4–14 migraine headache days per month) (25). The overall change from baseline in the number of MHDs during the study was significantly different for the 120 mg (−4.3 [95% CI −4.9, −3.7], p = 0.02) and 300 mg (−4.3 [95% CI, −4.9, −3.7], p = 0.02) galcanezumab dose groups compared with placebo (−3.4 [95% CI, −3.8, −2.9], p = 0.02). Overall, TEAEs were similar across all groups and no SAEs deemed related to the study drug occurred. The most commonly reported TEAE ( > 5%) across the various dose groups occurring more frequently as compared to placebo was injection site pain, which was self-limited.
In a pivotal phase 3 double-blind, randomized, placebo-controlled trial (EVOLVE-1), 1671 adult patients with episodic migraine were randomized 2:1:1 to monthly subcutaneous injections of placebo, galcanezumab 120 mg, or 240 mg over a 6-month period (NCT02614183) (26). Patients had 4–14 migraine headache days (MHDs) per month and at least two migraine attacks per month. Concurrent preventive medications were not permitted. Both dose groups significantly reduced the number of monthly MHDs over the 6-month study period by 4.7 days (120 mg) and 4.6 days (240 mg) compared with placebo (2.8 days; both dose groups p < 0.001). All key secondary objectives were also significant after multiplicity adjustment. There were no significant differences between the two doses of galcanezumab on any efficacy measures.
Significantly more patients in both galcanezumab dose groups experienced a 50%, (120 mg, 62.3%; 240 mg, 60.9%, placebo 42.5%; both dose groups p < 0.001) 75% (120 mg, 38.8%; 240 mg, 38.5%, placebo 19.3%; both dose groups p < 0.001), and 100% (120 mg, 15.6%; 240 mg, 14.6%, placebo 6.2%; both dose groups p < 0.001) reduction in MHDs over the 6-month trial. Overall, there were no significant or clinically meaningful differences in treatment emergent adverse events. A statistically significant, but not clinically meaningful mean decrease in diastolic blood pressure (DBP) was observed in the galcanezumab 240 mg dose group compared with placebo at month 6. Treatment-related discontinuation due to adverse events was less than 5% across all treatment groups and there were no SAEs considered to be related to study treatment. Anti-drug antibodies were present at baseline, 5.9% of patients in the placebo group, 8.9% (120 mg group) and 10.8% (240 mg group) in the galcanezumab dose groups. The percentage of patients with post-dose anti-drug antibodies was 1.7% (placebo), 3.5% (120 mg), and 5.2% (240 mg).
In EVOLVE-2 (NCT02614196), the second pivotal phase 3 randomized placebo-controlled trial in patients with episodic migraine, the baseline mean number of monthly MHD was similar to EVOLVE-1 (9.1) (26). As in EVOLVE-1, both galcanezumab dose groups (120 mg and 240 mg) demonstrated a significant improvement compared with placebo for overall mean change in monthly MHD (placebo −2.28; 120 mg −4.29; 240 mg −4.18; p < 0.001). The percentage of patients with MHD reductions of ≥ 50%, ≥ 75%, or ≥ 100% were also significantly higher for each galcanezumab dose group compared with placebo (p < 0.001).
REGAIN (NCT02614261) was a Phase 3, double-blind, randomized, placebo-controlled study in 1113 adult patients with chronic migraine who were randomized 2:1:1 to subcutaneous injections of placebo, galcanezumab 120 mg or galcanezumab 240 mg given once monthly for 3 months (27). The mean number of monthly MHDs at baseline was similar across treatment groups (19.4). Both galcanezumab doses demonstrated a significant difference from placebo in overall mean reduction (least square [LS] mean change) in number of monthly MHD during the 3-month double-blind treatment phase: Placebo −2.74, 120 mg −4.83, 240 mg −4.62 (p < 0.001 for each dose). Statistically significant improvements in MHD for both galcanezumab doses were observed at each month starting from month 1. The mean percentages over all 3 months of patients with 50% reduction from baseline in MHD were significantly higher for both GMB doses than placebo (p < .001 for both doses). Compared with placebo, patients in the 240 mg group also had significantly higher percentages of patients with 75% response rates (p < 0.001). There were no clinically meaningful differences between either GMB dose and placebo on any safety parameters except for a higher incidence of injection site reaction (p < 0.05), injection site erythema (p < 0.01), and sinusitis (p < 0.05) in the 240 mg group relative to placebo.
In a recently reported integrated subgroup analysis from the EVOLVE-1, EVOLVE-2, and REGAIN trials to determine whether there is a differential treatment effect in patients who had failed one and two previous preventive treatments vs. those who did not, galcanezumab 120 mg/240 mg significantly improved (p < 0.001) overall mean reduction of monthly MHD compared with placebo in both subgroups of patients (28). For the subgroup who failed prior preventive drugs, MHD reductions in EVOLVE studies were: placebo, −0.81; 120 mg, −3.45; and 240 mg, −3.85. In the REGAIN trial, MHD reductions were −1.44 for placebo, −5.91 for 120 mg, and −3.30 in the 240 mg dose group. Significant treatment-by-subgroup interactions were seen for the 240 mg (EVOLVE studies) and the 120 mg (REGAIN), suggesting better efficacy compared with placebo for these doses in patients who failed prior preventives.
Clinical implications
The emergence of a-CGRP mAbs as effective, safe, and well tolerated preventive treatment options for patients with episodic and chronic migraine has the potential to change paradigms on the approach and management of patients with migraine. Until now, the approach to drug prevention for migraine has been guided by well-established principles of “start low, go slow, and be patient”. At present, the principles for oral preventive medications include starting at the low dose, titrating slowly over weeks to months to a usual effective dose or until intolerable side effects develop, and waiting for 2–3 months after the target dose is achieved for efficacy (29–32). Currently available oral preventive drug options are associated with limited efficacy and side-effects that occur in a substantial proportion of patients. These considerations limit titration to a level that may provide efficacy, often necessitating their discontinuation. These factors may explain why adherence to oral preventive drugs over the course of 12 months after initiation occurs in less than 20% of patients (33–36).
Attributes of a-CGRP monoclonal antibodies relative to currently available oral migraine preventive drugs.

Adapted from: Israel H, Neeb L and Reuter U. CGRP Monoclonal antibodies for the preventative treatment of migraine. Curr Pain Head Rep 2018; 22: 38.
Selected outcomes of phase 2 and phase 3 trials of a-CGRP monoclonal antibodies for the preventive treatment of episodic and chronic migraine.
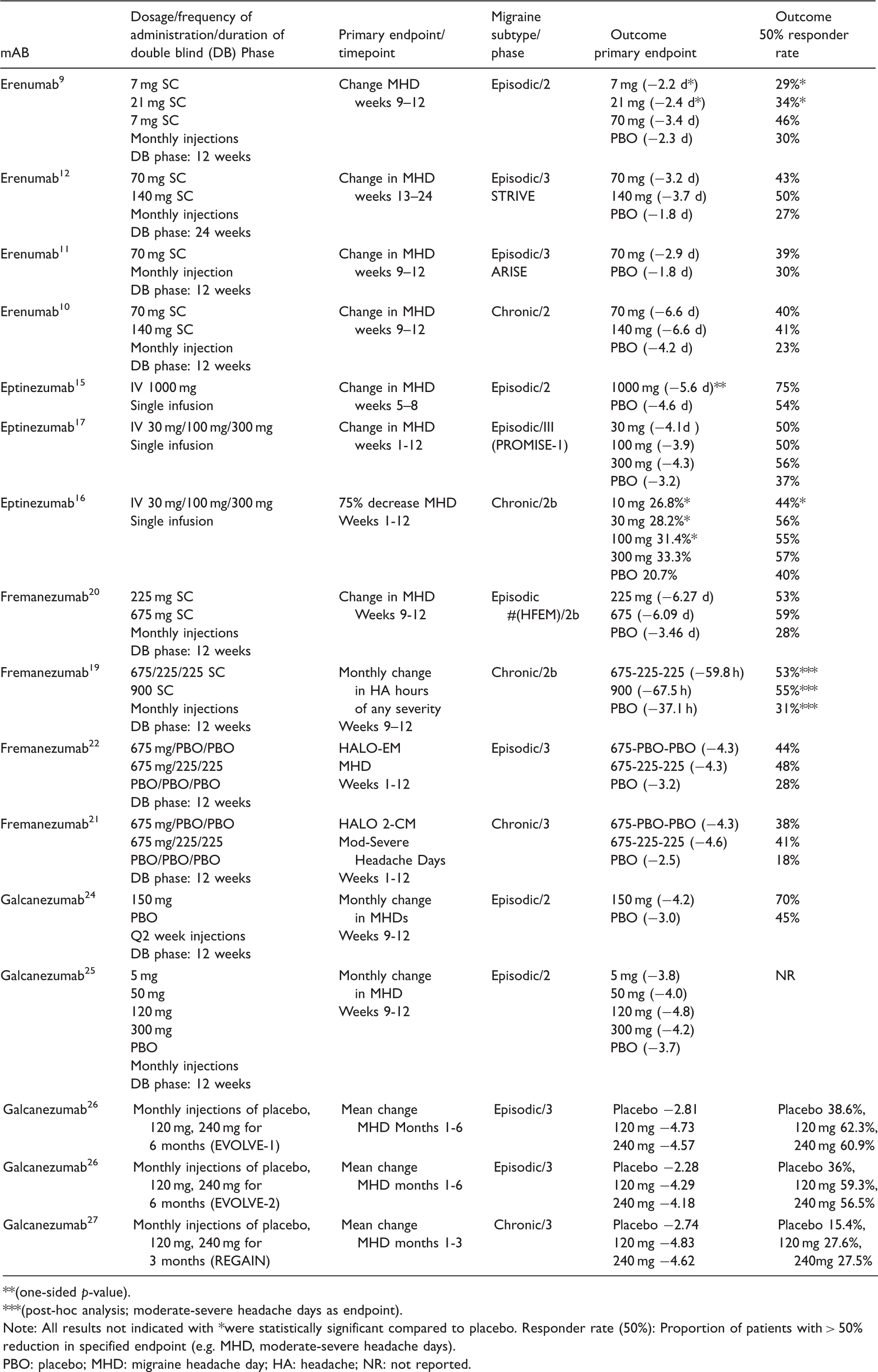
**(one-sided p-value).
***(post-hoc analysis; moderate-severe headache days as endpoint).
Note: All results not indicated with *were statistically significant compared to placebo. Responder rate (50%): Proportion of patients with > 50% reduction in specified endpoint (e.g. MHD, moderate-severe headache days).
PBO: placebo; MHD: migraine headache day; HA: headache; NR: not reported.
The large size and hydrophilic nature of mAbs results in minimal penetration through the blood-brain barrier. The blood-brain barrier, which functions at the level of endothelial cell tight junctions, is generally impermeable to water-soluble compounds with a molecular weight that exceeds 500 Daltons. A-CGRP mAbs far exceed this size and therefore, under normal conditions, their passage is expected to be negligible. In light of the fact that CGRP and its receptor is ubiquitous throughout the neuraxis, including both cortical and subcortical structures, the potential for a-CGRP mAbs to cross and access central sites is a concern. There are situations or clinical circumstances where the blood-brain barrier is disrupted; unusual situations such as aseptic or septic meningitis, and more common situations such as concussion. In these situations, the safety of CGRP monoclonal antibodies is unclear.
Another anatomic site where passage and access have unclear consequences is in the case of pregnancy. One of the distinct therapeutic advantages of a-CGRP mAbs – its long biological half-life – complicates its use to some extent in women of childbearing potential, especially since unintended pregnancies account for approximately 49% of all pregnancies and fetal exposure in these circumstances may occur. Cases of accidental exposure are certainly likely to occur in clinical practice, since women of childbearing potential account for the large majority of patients seen in the clinical practice of headache medicine and will represent a major target patient population for these therapeutic antibodies.
In animal models, circulating CGRP levels increase during pregnancy up to the time of delivery, and the responsiveness of vascular beds and uterine relaxation to CGRP appears to increase over the course of pregnancy (41). CGRP may therefore play a role in regulating uteroplacental blood flow and myometrial and uterine relaxation. CGRP may also play a role in maintaining normal gestational blood pressure and its blockade may increase the potential for gestational hypertension, pre-elcampsia, and eclampsia, the latter of which is already more prevalent in pregnant women with a history of migraine (42–44). At present, most available therapeutic monoclonal antibodies for the treatment of inflammatory and autoimmune diseases carry an FDA pregnancy rating of category B or C. IgG antibodies cross the placenta through the neonatal Fc receptor (FcRn) in syncytiotrophoblast cells, and during the first 20–22 weeks of pregnancy FcRn is absent and therefore there is minimal active transfer of IgG across the placenta (45). Placental transport, however, occurs progressively over the remaining course of pregnancy. CGRP has been demonstrated to be a potent vasodilator in the human placenta and appears to be important for uteroplacental blood flow and fetoplacental development (46). Fetal exposure to a-CGRP mAbs may occur during these periods, but the effect of CGRP blockade in a developing fetus is unknown. In a rat animal model, exposure to CGRP receptor antagonist CGRP8-37 in a dose-dependent fashion resulted in elevated maternal blood pressure, a reduction in pup weight, and increased fetal mortality rates (47). Clearly, clarifying the risk to pregnant mothers and fetus upon exposure to a-CGRP mAbs is of major importance, and prospective pregnancy registries with active surveillance should be established to bring answers to clinicians and their patients as quickly as possible. Until then, deliberate family planning and use of adequate contraception must be a part of the conversation between health care providers and their patients when discussing the potential use of a-CGRP mAbs for migraine prevention.
Summary
A-CGRP ligand and receptor mAbs represent a long-awaited disease-specific and mechanism-based treatment for the prevention of migraine. That not a single phase 2 or phase 3 trial has failed to reach its primary endpoint points to the efficacy of these biologics and the validity and sensitivity of the target. The unique attributes of these mAbs compared to established oral preventive drugs, including a tolerability profile that resembles placebo, has the potential to improve adherence and long-term outcomes and irrevocably alter the approach that clinicians take in counseling, educating, and managing patients with migraine. The preliminary evidence suggesting that these monoclonal antibodies are effective, potentially addressing current unmet needs, has an important bearing on patient access in the future when they are approved. While favorable long-term outcomes and safety have been reported, longitudinal surveillance is necessary when these drugs are exposed to heterogeneous patient populations. In addition, their safety in women will require active and deliberate surveillance in pregnancy registries. The effectiveness of existing (triptans) and emerging (CGRP receptor antagonists) acute therapies in those using a-CGRP mAbs, especially given overlapping mechanism(s) of action, will require future study.
Article highlights
Anti-CGRP monoclonal antibodies have been shown to be effective and well tolerated for the preventive treatment of episodic and chronic migraine, including patients who overuse acute medications, have failed prior preventive drug therapy, or are on concurrent preventive drugs. Anti-CGRP monoclonal antibodies have distinct advantages over currently available oral preventive drugs including very favorable tolerability profiles, lack of need for dose titration, swift onset of effect, convenient monthly or quarterly dosing schedule, progressive benefit over time, and low adverse event-related discontinuation rates in clinical trials. Long-term safety will require broad exposure in heterogenous patient populations in clinical practice.
Footnotes
Declaration of conflicting interests
The author declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Consulting: Amgen, Alder, Allergan, Novartis, Teva, Eli Lilly, Autonomic Technologies, Biohaven, eNeura, Neurolief, Zosano, WL Gore, Vedanta Associates, Promius Pharma, Nocira,, Electrocore, Ipsen, Impel, Satsuma, Theranica. Compensation for activities related to data safety monitoring committee from Axsome. Compensation related to CME content development: Healthlogix, Medicom Worldwide, Medlogix Communications, MedNet, Miller Medical Communications, PeerView Operation Services America, Web MD/Medscape, American Academy of Neurology, American Headache Society, PeerView Institute for Medical Education, Chameleon Communications, Academy for Continued Healthcare Learning, Universal Meeting Management, Haymarket Medical Education, Global Scientific Communications, UpToDate, Meeting LogiX. Royalties from editorial or book publishing: Oxford University Press, Cambridge University Press, Wiley Blackwell, Sage, Wolters Kluwer Health. Consulting use agreement through employer: NeuroAssessment Systems, Myndshft. Equity: Aural Analytics, Healint, Theranica, Second Opinion/Mobile Health, Epien, King-Devick Technologies, Ontologics.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.