
Abstract
Background
Recently, the adenosine triphosphate (ATP) sensitive potassium channel opener levcromakalim was shown to induce migraine attacks with a far higher incidence than any previous provoking agent such as calcitonin gene-related peptide. Here, we show efficacy of ATP sensitive potassium channel inhibitors in two validated rodent models of migraine.
Methods
In female spontaneous trigeminal allodynic rats, the sensitivity of the frontal region of the head was tested by an electronic von Frey filament device. In mice, cutaneous hypersensitivity was induced by repeated glyceryl trinitrate or levcromakalim injections over nine days, as measured with von Frey filaments in the hindpaw. Release of calcitonin gene-related peptide from dura mater and trigeminal ganglion was studied ex vivo.
Results
The ATP sensitive potassium channel inhibitor glibenclamide attenuated the spontaneous cephalic hypersensitivity in spontaneous trigeminal allodynic rats and glyceryl trinitrate-induced hypersensitivity of the hindpaw in mice. It also inhibited CGRP release from dura mater and the trigeminal ganglion isolated from spontaneous trigeminal allodynic rats. The hypersensitivity was also diminished by the structurally different ATP sensitive potassium channel inhibitor gliquidone. Mice injected with the ATP sensitive potassium channel opener levcromakalim developed a progressive hypersensitivity that was completely blocked by glibenclamide, confirming target engagement.
Conclusion
The results suggest that ATP sensitive potassium channel inhibitors could be novel and highly effective drugs in the treatment of migraine.
Abbreviations:
ATP: adenosine triphosphate; cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate; CGRP: calcitonin gene-related peptide; DMSO: dimethyl sulfoxide; EIA: enzyme immunoassay; GTN: glyceryl trinitrate; KATP: ATP-sensitive potassium; KIR: inwardly-rectifying potassium channels; NO: nitric oxide; PACAP: pituitary adenylate cyclase activating peptide; PKA: protein kinase A; PKG: protein kinase G; sAC: soluble adenylyl cyclase; SD: Sprague Dawley; sGC: soluble guanylyl cyclase; SIF: synthetic interstitial fluid; STA: spontaneous trigeminal allodynia; SUR: sulfonylurea receptor; TG: trigeminal ganglia
Introduction
Migraine affects one billion people worldwide. As a cause of disability, it is number two surpassed only by low back pain according to the Global Burden of Disease study (1). It has an estimated economic cost of €50 – €111 billion per year in Europe (2).
Treatment possibilities have expanded, most recently with the advent of antibodies directed against calcitonin gene-related peptide (CGRP) or its receptor. Small molecule CGRP receptor antagonists recently reached the market. However, none of these are effective in more than 50–60% of patients (3). Thus, the unmet need for better migraine treatments is still huge.
Potassium channels are found in most living organisms, where they serve a variety of crucial functions. Within the trigeminovascular system, a number of K+ channel subtypes such as two-pore potassium channels (K2Ps), big calcium activated potassium channels (BKCa) and ATP-sensitive potassium (KATP) channels have been implicated in the aetiology of migraine (4–7). KATP channels are expressed in various tissues and serve to couple cellular metabolism, via adenine nucleotides to membrane potential and excitability (8–11). This enables KATP channels to regulate a diverse range of physiological processes including maintenance of blood glucose homeostasis, control of excitation-contraction coupling in muscular tissue, and regulation of vascular tone (12–14).
With the use of a human migraine model, it was shown that the KATP channel opener levcromakalim appears to be a potent migraine trigger (15). The use of this model has proven beneficial in identifying signaling molecules, such as nitric oxide (NO) (16–18), pituitary adenylate cyclase activating peptide (PACAP) (19,20) and CGRP (21), with a role in migraine pathophysiology (22). Thus, migraine provocation by KATP channel opening immediately suggests the possibility that inhibitors of KATP channels may be effective in migraine. However, that a naturally occurring substance or mechanism can induce migraine does not necessarily mean that their reciprocal modulators are effective in the treatment of migraine attacks (23,24).
There are no human studies of KATP channel inhibitors in relation to migraine treatment. Therefore, it is pertinent to obtain as good evidence of efficacy in animal models as possible before recommending industry to pursue KATP channel inhibition as a drug target. Here, we have used two validated rodent models of migraine. One model is a genetically inbred strain, the spontaneous trigeminal allodynic (STA) rat, which exhibits tactile hypersensitivity in the head but not in the hindpaws (25,26). The other model uses repeated injection of glyceryl trinitrate (GTN) to provoke a general state of hyperalgesia in mice (27,28). Both animal models respond to migraine-specific drugs including the 5-HT1B/D receptor agonist sumatriptan, the CGRP receptor antagonist olcegepant and a CGRP antibody (25–28). In addition, a validated ex vivo model of capsaicin-induced CGRP release from migraine-relevant tissue was used (29,30). Our results show that KATP channel inhibition is a relevant future target for novel migraine treatments.
Materials and methods
Animals
Female STA rats (body weight 216–255 g; age 19 weeks, n = 24) were derived from a single breeding pair originally sourced from the Thomas Jefferson University under license (26) and subsequently bred in house. Periorbital thresholds for this breeding pair, measured via manual von Frey filament testing, have been described previously (25). For experiments with control rats we used wild type Sprague-Dawley rats (body weight 196–280 g; age 8–13 weeks, n = 22) obtained from Charles River (Sulzfeld, Germany). All rats were group housed in Tecniplast 1354G Eurostandard type IV polycarbonate cages (L × W ×H: 60 × 38 × 20 cm; Brogaarden, Denmark) using a 12-h light/dark cycle with lights on at 4 am. Individual opaque red polycarbonate shelters (20 ×11.5 × 16 and 15 × 9 × 9 cm respectively), together with an aspen biting stick (10 × 2 × 2 cm; Tapvei, Estonia) and piece of hemp rope suspended from the cage lid were provided in each homecage for retreat and enrichment purposes. Bedding consisted of Enviro-Dri nesting material (Brogaarden, Denmark). Standard rat chow (Altromin) and tap water were available ad libitum in the animals’ home cage environment. Humidity ranged from 45–65%. All in vivo experiments were performed in a laboratory in the animal housing facilities between 8 am and 4 pm.
When cross-over studies were performed, at least 3–4 days were allowed for drug washout before rats were allocated to a different treatment. Importantly, low periorbital threshold responses at baseline was confirmed prior to treatment. Experiments were approved by the Danish Animal Experiments Inspectorate and performed in accordance with the relevant guidelines and regulations of approval number 2014-15-0201-00256.
Male C57Bl/6J mice (Taconic, Denmark) of 8–9 weeks (22–31 g) at start of experiment were used following 7 days acclimatisation to the animal facility. In total, 107 mice were used to complete the study. Mice were housed in IVC type II cages (Tecniplast) in groups of six with ad libitum access to food (Altromin 1314, Altromin GmbH & Co., Seelenkamp, Germany) and tap water. The cages were floored with sawdust (Aspen chip, Tapvei, Finland) and enriched with a red-tinted polycarbonate shelter (Molytex, Denmark), nesting material (Happi-Mats, Scanbur, Denmark) and biting sticks (Tapvei, Finland). The light cycle was 12/12, light on at 6 am. All handling of animals was done between 7.30 am and 4 pm in a dedicated procedure room. The experiments were approved by the Danish Animal Experiments Inspectorate and performed in accordance with the relevant guidelines and regulations of approval number 2017-15-0201-01358.
In both species, one experimental unit represents one animal.
Measurement of glucose levels
The rat was gently restrained by one investigator, while another investigator used a 1 ml plastic insulin syringe (29 gauge) to obtain a venous blood sample (approximately 50 µl) from the dorsal aspect of the hindpaw. A new syringe was used for each blood sample. Blood glucose was measured immediately according to the manufacturer’s instructions using the Accu-Chek, Aviva kit (Roche Diabetes Care, Mannheim, Germany). Blood glucose was either measured in a separate group of rats or at the end of the experiment once all behavioral testing was completed. Accordingly, there would have been no effect of the blood sampling protocol on the sensory threshold responses.
Periorbital pain thresholds in STA rats
The sensitivity of the frontal region of the head (V1 ophthalmic trigeminal dermatome), to static mechanical stimulation was measured using an electronic von Frey device fitted with a rigid plastic tip (IITC LifeScience Inc, USA). The rat was gently restrained using a cotton towel and held in a prone position in the lap of the investigator with its head and neck region left exposed and unrestricted. The tip of the device was then applied with increasing force (maximum of 450 g) to the right periorbital area above the eyes until the rat withdrew its head, laterally rotated its head and/or vocalized. The soft tissue around the eye was carefully avoided. The process was then repeated over the midline, the left and then the right periorbital areas above the eye. The average of these latter three measurements was considered the withdrawal threshold (g). If a stimulus had to be reapplied due to inappropriate application, care was taken not to apply the probe tip to the exact same location within the region being measured. The whole procedure typically took less than 2 minutes to perform.
Hindpaw pain thresholds in STA rats
Hindpaw sensitivity to mechanical stimulation was assessed using an electronic Randall Selitto paw pressure device (IITC LifeScience Inc, USA) as described previously (25). Briefly, the rat was gently restrained within the hand and lower arm of the investigator and the right hindleg extended and the hindpaw inverted. The probe tip was then placed on the paw between the foot pads and increasing force uniformly applied (maximum of 450 g) until the rat withdrew the hindpaw and/or vocalized. The average of three measurements obtained from defined parts of the hindpaw excluding the footpads was considered the paw pressure withdrawal threshold (g).
To assess hindpaw thresholds to noxious thermal stimulation, we used a hot/cold plate (IITC LifeScience Inc, USA) with a pre-set plate temperature of 48°C. The latency (s) to respond to the thermal stimulus which consisted of either licking, rapid shaking, or stepping of the hindpaws, was recorded from the moment the rat was placed onto the surface of the plate. A cut-off time was set at 60 sec. No rats showed signs of thermally-induced damage to the paws throughout the duration of the study.
Assessment of motor function in rats
To assess if motor function was putatively impaired by drug treatment, for each rat a simple observational analysis was made according to methods modified from Zahn and co-workers (1997) and Hao & Xu (1996) (31–33). The rat was placed onto a clean tabletop and hindlimb gait scored as (2, normal; 1, moderate hindlimb weakness/splaying; 0, complete hindlimb weakness/splaying). The ability to walk was then scored as (2, normal; 1, walks with mild deficit; 0, no attempt to walk). Next, the dorsum of the right hindpaw was drawn across the edge of the table, a stimulus which elicits a lifting of the paw onto the surface of the table and scored as (2, normal; delay of 1–2 s, 1; 0, delay >2 s). Finally, the whole-body righting reflex was assessed by placing the rat horizontally with its back on the table which normally gives rise to an immediate, coordinated twisting of the body to an upright position (2, normal; delay of 1–2 s, 1; 0, delay >2 s). Each test was performed once, and the complete battery of tests took less than 1 minute to perform for each rat. The total score was summed for each rat and statistical comparisons between treatment groups made using a non-parametric ANOVA.
Induced hindpaw hyperalgesia in mice
Sensitivity towards mechanical stimulation to a series of von Frey filaments (Ugo Basile, Italy) was evaluated at the plantar surface of the left hindpaw using the up-down test paradigm (34). Analysis of the data was performed with an improved algorithm for calculation of 50% thresholds based on exact between-filament intervals (35). Filaments were applied until withdrawal of the foot or up to 3 sec in case of non-withdrawal. Rapid removal of the paw during or in relation to the end of stimulation was considered a positive response. For testing, animals were kept in individual clear plexiglass cages (L × W × H: 10 × 10 × 12 cm) with a mesh floor (IITC Life Science, US) (27). Measures were taken before and after drug administrations.
GTN and levcromakalim were administered every other day for a total of five times. Using this paradigm, GTN mice present hindpaw and cephalic hyperalgesia that becomes more severe with every injection (27,28). We tested if glibenclamide could prevent the development of GTN-induced hyperalgesia by injecting glibenclamide 15 min prior to GTN on each of the five test days. The same protocol was applied to investigate if levcromakalim, like GTN, could induce hyperalgesia. First, we did a small experiment for dose-finding purposes and in a subsequent experiment the effect of levcromakalim was antagonised by prior treatment with glibenclamide.
All pharmacology experiments were performed with the investigator blinded to treatment, with rats and mice randomly allocated to vehicle or treatment groups.
CGRP release ex vivo
Female STA (215–318 g, n = 16) rats were used for ex vivo experiments. Origin of STA rats, housing and approval numbers by the Danish Animal Experiments Inspectorate are the same as described above. On the day of experiment, the rats were deeply anaesthetised by i.p. injection of a mixture of pentobarbital (90 mg/kg) and lidocaine (9 mg/kg) followed by decapitation and dissection of tissues as previously described in detail (29,36). Briefly, the skull was cut mid-sagittally and the brain halves were carefully removed while the dura mater was left attached to the skull. Then the skull halves (with intact dura mater and without the trigeminal ganglion (TG)) and the carefully dissected TG were immersed in 25 ml synthetic interstitial fluid (SIF) at room temperature and repeatedly washed (every 5 min) for 30 minutes with SIF (in mM: 108 NaCl, 3.5 KCl, 3.5 MgSO4, 26 NaHCO3, 11.7 NaH2PO4, 1.5 CaCl2, 9.6 Na+ gluconate, 5.6 glucose and 7.6 sucrose).
The two TGs from each rat were individually placed in 350 µl SIF in the cap of an Eppendorf tube and the two skull halves were placed on separate platforms. The tissues were then placed in a humid chamber at 37ºC in an incubator. They were washed with SIF five times without touching the tissue. The basal CGRP release was determined after 10 minutes of incubation in SIF after the fifth wash.
The experiments were performed according to two different protocols, as shown in Figure 1. In the first protocol, sampling was performed after each 10 min incubation with increasing concentrations of capsaicin (10 nM to 10 µM) or its vehicle. In the second protocol, the tissues were incubated for 10 min with vehicle or 3 µM glibenclamide; after sampling, the tissues were incubated with a mixture of 1 µM capsaicin and 3 µM glibenclamide. After sampling of each concentration, the residual SIF was discarded and new SIF containing the next concentration of capsaicin or vehicle was added.

Schematic overview of experimental protocol. The upper panel shows the experimental protocol for concentration-response curves of CGRP release after administration of capsaicin in increasing concentrations. The lower panel represents the experimental protocol for studies investigating the effect of glibenclamide on capsaicin-induced CGRP release. The hemisected skull with dura and TG from one side served as control to the treatment in the skull and TG from the other side.
For measurement of CGRP, samples were processed with commercial EIA (Enzyme–linked immunoassay) kits (SPIbio, Paris, France). The antibody in the CGRP-EIA kit is directed against human-CGRP but has 120% cross-reactivity to rat CGRP. The CGRP detection level is about 0.7 pg/ml. Previous studies have shown that the EIA assay is strictly specific for the entire sequence of CGRP, making no cross reactivity with structurally matched peptides; that is, calcitonin, amylin or with CGRP degradation fragments. We have matched the signal by adding the known concentration of synthetic α-CGRP in this assay. The protocol provided with the kit was followed in detail. The optical density was measured at 410 nm using a micro plate photometer (Tecan, infinite M200, software SW Magellan v.6.3).
Drugs
For use in rats, buprenorphine (Indivior UK, Temgesic 0.3 mg/ml) was diluted to 0.1 mg/ml in saline and administered s.c. 0.1 mg/kg. Glibenclamide (Tocris/Bio-Techne, UK) was dissolved to 0.1, 1 and 10 mg/ml in vehicle (20% dimethyl sulfoxide (DMSO)-50% alcohol (96%)-30% saline) and administered i.p. at 1 ml/kg. Suspensions of gliquidone (Sigma-Aldrich Denmark A/S, Denmark) were made to 0.5, 5 and 50 mg/ml in milli-Q water with 25% (w/v) 2-hydroxypropyl-beta-cyclodextrin and subsequently administered p.o. at 2 ml/kg.
For use in mice, glibenclamide (Tocris/Bio-Techne, UK) was diluted to 0.1 mg/ml in a 5% DMSO solution and administered at 1 mg/kg. Levcromakalim (Tocris/Bio-Techne, UK) was dissolved in DMSO and diluted in saline to the desired concentration for injection of 0.1, 0.5 and 1 mg/kg in a volume of 10 ml/kg resulting in a maximum concentration of DMSO of 2%. GTN 7.89 mg/ml in ethanol (Capital Region, Hospital Pharmacy) was diluted in saline to 1 mg/ml (12.8% ethanol) and administered at 10 mg/kg. All drugs were administered i.p. to mice in a volume of 10 ml/kg. For all drugs, the vehicle was appropriate to the injected drug.
For the ex vivo assay, capsaicin and glibenclamide were purchased from Tocris (Bio-Techne Ltd., Abingdon, UK). The stock of capsaicin was prepared in ethanol to 10 mM and was further diluted in SIF for working solutions. Glibenclamide was prepared in ethanol to a master stock of 1 mM and was further diluted in SIF to 3 µM, as used in the experiments.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 8 (Graph Pad Software Inc., San Diego, CA). Group sizes for the pharmacology experiments on cutaneous threshold values were estimated as a function of the desired effect size (approximately 50% change vs. corresponding vehicle treatment) where we assumed a significance level of 5% and a power of 90% (37). All data are presented as mean ± SEM, p < 0.05 was considered statistically significant.
Rat behavioral experiments were performed as cross-over experiments where the individual rat received all treatments on different test days in a randomized manner. Repeated measure analysis of variance (ANOVA) or mixed effects model for data sets with missing values were used to analyze the overall effects of treatments followed by Bonferroni’s multiple comparisons test. For comparisons between only two groups/treatments, paired or unpaired t-tests were performed as appropriate. Mouse 50% withdrawal thresholds were square root transformed to obtain normally distributed data, and data were analyzed by repeated measure ANOVA followed by Bonferroni’s post hoc test for comparison between treatment groups. For in vitro assays, the release of CGRP (measured as immunoreactive CGRP in the samples) was expressed in pg/ml, given as mean values ± SEM. Absorbance was recorded, and values were calculated through an interpolation method using an equation derived from the standard curve. The value of the diluted samples was multiplied by 2. Paired data (control site and test site) were analyzed for overall effect of treatment by two-way ANOVA, one-way ANOVA or mixed effects analysis for data sets with missing values followed by Bonferroni’s multiple comparisons test.
Results
KATP channel inhibition in STA rats
The non-selective KATP channel inhibitor glibenclamide was first tested in STA (spontaneous trigeminal allodynia) rats and blood glucose levels measured to determine doses of glibenclamide that actively engage pancreatic KATP (e.g. Kir6.2/SUR1) channels. Accordingly, glibenclamide (0.1–10 mg/kg, i.p.) treatment lowered blood glucose levels (F(4,20) = 116.1, p < 0.0001, n = 4–6 per group). Note that we observed that the applied vehicle (20% DMSO-50% alcohol (96%)-30% saline) increased blood glucose levels compared with baseline plasma glucose levels in non-injected naïve rats (Figure 2(a)). The 2 h post-treatment analysis revealed that the 1 mg/kg dose of glibenclamide lowered blood glucose levels by 55% compared with vehicle treatment (p < 0.001) and this was the lowest active dose of glibenclamide.
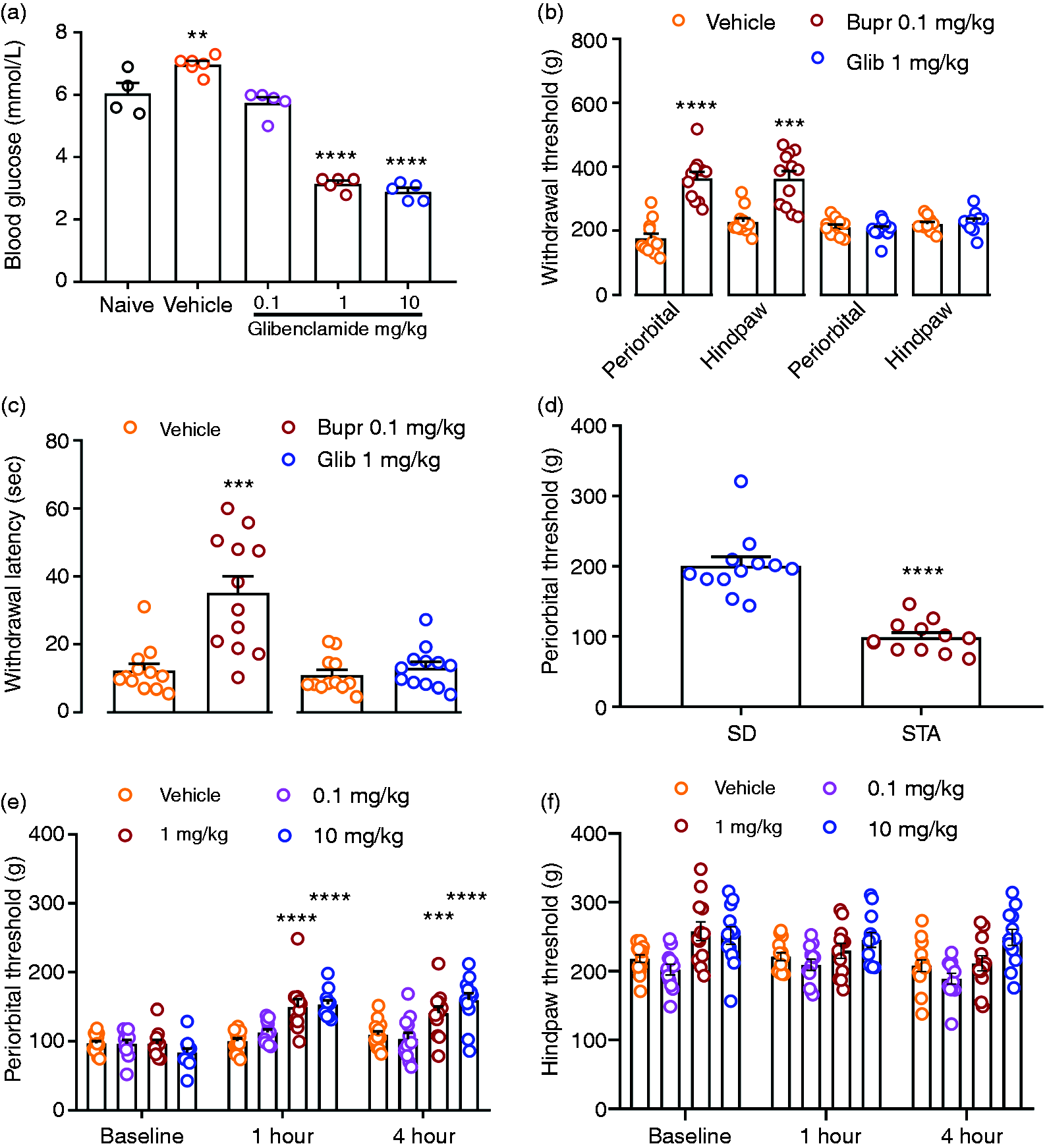
Effects of glibenclamide on blood glucose and sensory nociceptive thresholds. (a) Effects of glibenclamide (0.1–10 mg/kg, i.p.) on blood glucose levels (mmol/l) in four to six STA rats. (b) Cutaneous periorbital and hindpaw withdrawal thresholds (g) were increased after buprenorphine (0.1 mg/kg, s.c.; n = 12), but not after glibenclamide (1 mg/kg, i.p.; n = 10) in wild type Sprague Dawley (SD) rats. (c) Latency responses (s) to thermal hindpaw stimulation after buprenorphine 0.1 mg/kg (n = 12) and glibenclamide 1 mg/kg (n = 12) as compared to vehicle treatment in SD rats. (d) Periorbital baseline withdrawal threshold response to cutaneous mechanical stimulation in SD (n = 12) and STA rats (n = 12). Thereafter, glibenclamide (0.1–10 mg/kg, i.p.; n = 12–15) was administered and the effects on (e) periorbital and (f) hindpaw post-treatment thresholds were evaluated. Data represent mean ± SEM.
Next, we assessed the effects of glibenclamide on nociceptive thresholds in naïve Sprague-Dawley (SD) rats. Systemic administration of glibenclamide (1 mg/kg i.p.) had no effect on cephalic or hindpaw thresholds in response to cutaneous mechanical stimulation (p = 0.541 and p = 0.564 vs. vehicle, respectively, n = 10), (Figure 2(b)). In contrast, the partial µ-opioid receptor agonist buprenorphine increased both cephalic and hindpaw thresholds by 107% and 59% (p = 0.0001 and p = 0.001 vs. vehicle respectively, n = 12). Similarly, glibenclamide had no effect on thermal nociceptive thresholds in naïve rats whereas buprenorphine administration increased the latency response to thermal stimulation by 185% (p = 0.001 vs. vehicle, n = 12), (Figure 2(c)). Thus, glibenclamide has no effect on normal nociceptive transmission.
Figure 2(d) shows that periorbital thresholds to cutaneous mechanical stimulation were 51% lower in STA rats compared with control SD rats (p < 0.0001, n = 12), indicative of sensitization of trigeminal nociceptive signalling in STA rats. To better understand a potential role for KATP channels in pain transmission relevant to migraine, we administered glibenclamide to female STA rats. Glibenclamide (0.1–10 mg/kg, i.p.) increased the periorbital threshold to mechanical stimulation (treatment × time interaction, F(6, 57) = 8860, p < 0.0001) in STA rats (Figure 2(e)). Post-treatment analysis revealed that this effect was significant for both the 1 and 10 mg/kg doses of glibenclamide (p < 0.001 vs. vehicle, n = 12–15) at both the 1 and 4 h timepoints post-treatment. Notably, glibenclamide had no effect on the normosensitive hindpaw threshold to mechanical stimulation when comparing to vehicle treatment responses at the same time-points (treatment × time interaction, F(3.7, 35.2) = 1572, p = 0.2063, n = 12–15, thereby confirming specificity for trigeminal versus somatic pain circuits (Figure 2(f)).
Glibenclamide has been reported to be an agonist at human transient receptor potential ankyrin 1 channels in contrast to other sulphonylurea compounds (38). Thus, to help confirm that glibenclamide-mediated effects on cephalic threshold responses in STA rats were not simply compound specific, we assessed the effects of the structurally distinct KATP channel inhibitor gliquidone (39). A dose range finding pilot study on five rats showed a significant fall in glucose levels after treatment with 10 and 100 mg/kg p.o. of gliquidone of 40% and 63%, respectively. The lowest dose of 1 mg/kg only caused a non-significant 3% fall in glucose levels (p = 0.988). Therefore, we decided to continue with the two highest doses in the main experiment, in which 11 female STA rats were used. In these rats, gliquidone caused a significant (F(2,15) = 53.22, p < 0.0001) decrease in blood glucose levels of 43% versus vehicle in both the 10 mg/kg and 100 mg/kg groups (n = 11) (Figure 3(a)). Figure 3(b) illustrates that gliquidone (10–100 mg/kg, p.o.) caused a significant increase in periorbital threshold to mechanical stimulation when administered to STA rats (treatment × time interaction, F(2,23) = 3.77, p = 0.036). Gliquidone had no effect on hindpaw threshold to mechanical stimulation (Figure 3(c)), thereby confirming specificity for trigeminal versus somatic pain circuits. Finally, accumulated motor scores were tested in STA rats treated with glibenclamide (n = 12) and gliquidone (n = 12) as compared to vehicle responses in the same individual rats. In all STA rats and treatments, the accumulated motor score reached a maximum value of eight, indicating a normal motor function. This supports that the reversal of cutaneous hypersensitivity did not simply occur because of non-specific effects on efferent motor function.

Cephalic hypersensitivity in STA rats is attenuated by gliquidone. (a) Effects of gliquidone (10–100 mg/kg, p.o.) on blood glucose levels (mmol/l) in STA rats. Cutaneous (b) periorbital and (c) hindpaw threshold responses (g) to mechanical stimulation 2 and 4 h after treatment with the selective KATP channel inhibitor gliquidone (10–100 mg/kg, p.o.; n = 11). Data represent mean ± S.E.M.
KATP channel inhibition in mice
As previously described, repeated administration of GTN (10 mg/kg, i.p.) produced hindpaw and cephalic allodynia in mice (27,28,40,41). Treatment with glibenclamide (1 mg/kg, i.p.) 15 min before GTN on every test day (1, 3, 5, 7 and 9) resulted in significantly higher 2 h post-drug withdrawal thresholds in response to GTN than in mice administered vehicle before GTN treatment (overall effect of treatment, F(2,32) = 41.38, p < 0.0001) (Figure 4(b)). The basal response, measured before injection of GTN on each test day, was significantly (overall effect of treatment, F(2,32) = 39.54, p < 0.0001) reduced from the third test day (Figure 4(a)). Glibenclamide had no significant (p >0.05) effect on the basal responses found after GTN treatment (Figure 4(a)).

Hindpaw hyperalgesia in GTN-sensitized mice is attenuated by glibenclamide. Glibenclamide (1 mg/kg, i.p.) or vehicle (5% DMSO, 10 ml/kg, i.p.) was given 15 min prior to GTN (10 mg/kg, i.p.) on each test day (1, 3, 5, 7 and 9). The sensitivity to mechanical stimulation with von Frey filaments was measured as basal values before (a) and 2 h after GTN injection. (b) Data are square root transformed 50% withdrawal thresholds and depicted as mean ± SEM.
The 2 h post-drug response to the KATP channel opener levcromakalim dose-dependently (0.1–1 mg/kg, i.p.) induced hyperalgesia (Figure 5(b)). Whereas the acute hyperalgesia to GTN injection occurred after just a single injection, the effect of levcromakalim was not robust until the fifth test day (third dose). But, as observed for GTN-induced hyperalgesia, the response became stronger throughout the protocol (Figure 5(b)) (treatment effect F(3,20) = 9.71, p = 0.0004). The levcromakalim-induced hyperalgesia was most pronounced 2 h after administration (Figure 5(c)). The basal response to levcromakalim was significantly reduced (overall effect of treatment, F(3,20) = 2.82, p < 0.065) at the ninth test day in the two highest doses (0.5 (p = 0.025) and 1 mg/kg (p = 0.027)) (Figure 5(a)), but was reduced from the fifth day.

Hyperalgesia mediated by the KATP channel opener levcromakalim in mice. On every test day (1, 3, 5, 7 and 9) sensitivity to mechanical stimulation with von Frey filaments was measured at baseline and every hour for 4 h following injection of levcromakalim (0.1, 0.5 and 1.0 mg/kg, i.p.) or vehicle (2% DMSO, 10 ml/kg). (a) Baseline thresholds (g) measured before administration of levcromakalim. (b) Effect of levcromakalim on every test day measured 2 h after dosing. (c) Here, data are shown for test day 5 wherein the maximal hyperalgesia mediated by levcromakalim occurs 2 h after injection, thereby supporting the choice of this timepoint for subsequent experiments. Data are square root transformed 50% withdrawal thresholds (g) and depicted as mean ± SEM.
Based on this initial dose-finding experiment, we decided to assess the ability of glibenclamide to block levcromakalim (1 mg/kg, i.p.) induced hyperalgesia in a fully powered experiment (Figure 6). In this set of experiments, levcromakalim induced a significant basal hyperalgesia from the fifth day to the end of the experimental period on the ninth day (overall effect of treatment, F(3,44) = 24.55, p < 0.0001). This basal effect was on each of these days significantly (day 5, p = 0.0162; day 7 and 9, p < 0.0001) inhibited by glibenclamide (Figure 6(a)). We found that glibenclamide completely inhibited the development of 2 h post-drug hyperalgesia on every test day (overall effect of treatment, F(3, 44) = 24.93, p < 0.0001) (Figure 6(b)). This experiment also included a glibenclamide control group to test effects of this drug alone on hindpaw hyperalgesia. This group was not different from the vehicle control group (p >0.99 on all test days) (Figure 6(b)).

Hindpaw hyperalgesia in levcromakalim-sensitized mice is inhibited by glibenclamide. Levcromakalim (1 mg/kg, i.p.) or vehicle (2% DMSO, 10 ml/kg) was administered on every test day (1, 3, 5, 7 and 9) 15 min after an injection of glibenclamide (1 mg/kg, i.p.) or vehicle (5% DMSO, 10 ml/kg, i.p.). The sensitivity to mechanical stimulation was measured before (a) and 2 hrs after (b) levcromakalim dosing with von Frey filaments. Glibenclamide attenuated the basal hypersensitivity from the fifth day (a) and completely inhibited levcromakalim-induced hyperalgesia but had no effect on withdrawal thresholds on its own (b). Data are square root transformed 50% withdrawal thresholds (g) and depicted as mean ± SEM.
CGRP release from dura and TG
CGRP release has for a long time been suggested to be involved in the pathophysiology of migraine and the most recent treatment strategies of migraine (triptans) inhibit the release of CGRP or directly block the action of CGRP.
To find the most optimal concentration of capsaicin for the subsequent blockade experiments with glibenclamide, the release of CGRP was measured after cumulative application of capsaicin and its vehicle in tissue from four rats. The results revealed a significant concentration-dependent CGRP release from dura mater (F(4,22) = 39.67, p < 0.0001) and TG (F(4, 22) = 13.39, p < 0.0001) in relation to vehicle (Figure 7(b) and (c)). The maximum release of CGRP from dura mater was at 1 µM capsaicin, resulting in a 10-fold increase (n = 4) compared to vehicle (Figure 7(a)). In TG, the maximum response was measured at 10 µM capsaicin and increased 1.85-fold compared to vehicle (n = 4) (Figure 7(b)). Based on the above results, we performed blockade experiments with glibenclamide at a capsaicin concentration of 1 µM as it evoked a maximal or submaximal response in dura and TG, respectively. We used glibenclamide in a concentration of 3 µM, as it was previously shown to inhibit levcromakalim-induced rat basilar artery relaxation (42) and to sufficiently block KATP channels comprised of Kir6.2 and SUR1 subunits located in peripheral tissue (43,44). There was no significant difference in CGRP release between treatment with vehicle and 1 µM glibenclamide in the two tissues (dura p = 0.441; TG p = 0.881) (Figure 7(c) and (d)). Thus, glibenclamide had no effect on baseline CGRP release.
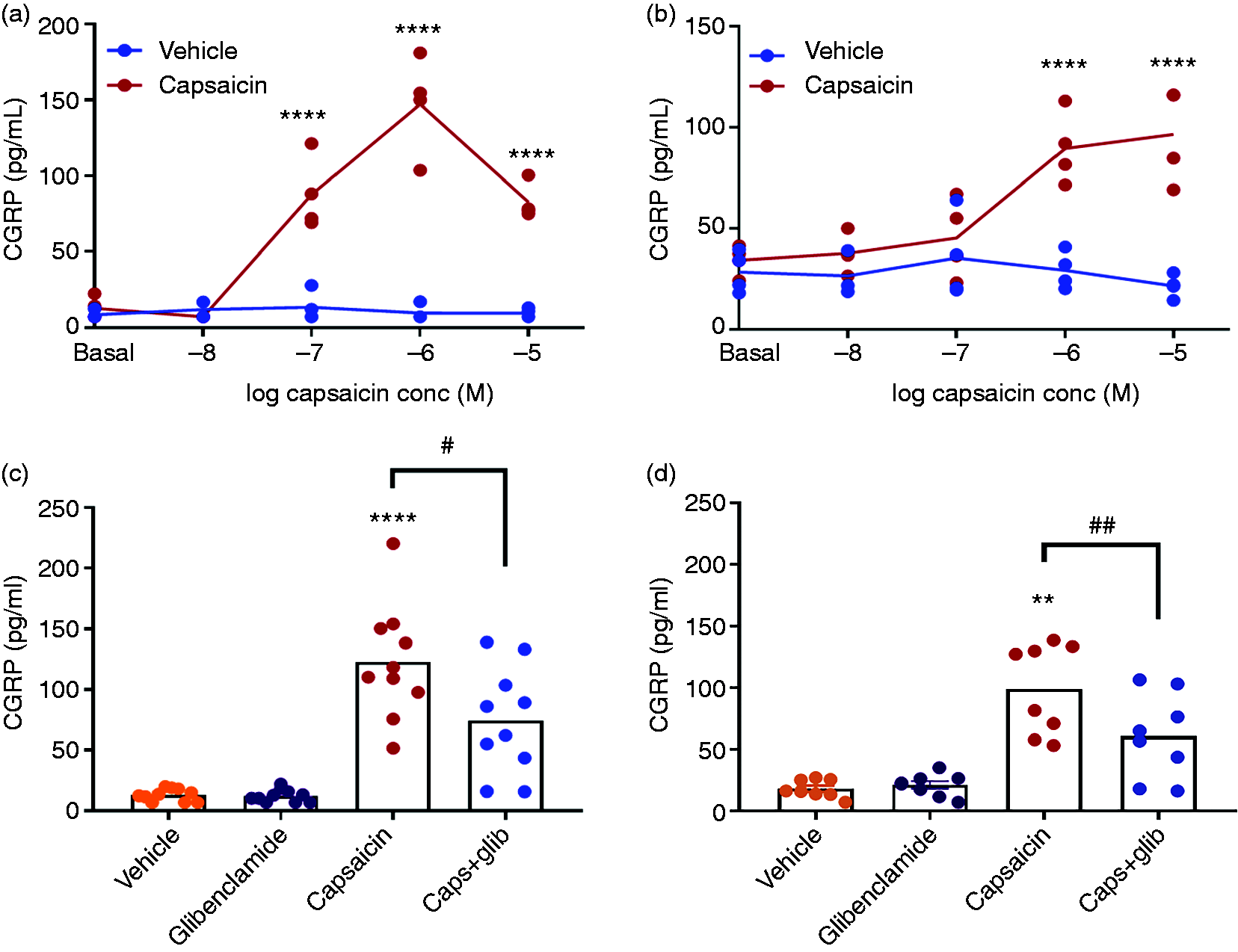
Ex vivo CGRP release is inhibited by glibenclamide. ((a) and (b)) CGRP release after increasing concentrations of capsaicin in dura mater and trigeminal ganglion (TG), n = 4. ((c) and (d)) The amount of CGRP released during 10 min incubation with vehicle, 1 µM capsaicin or 1 µM capsaicin + 3µM glibenclamide in (c) dura mater and (d) trigeminal ganglion from STA rats.
In dura mater, a single concentration of 1 µM capsaicin induced a significant (p < 0.0001) release of CGRP amounting to 123 + 15 pg/ml (n = 10). In the presence of glibenclamide, the capsaicin-induced CGRP release was significantly (p < 0.031) decreased by 40% of the response to capsaicin alone (Figure 7 (c)). In TG, 1 µM capsaicin significantly (p < 0.0011) increased the CGRP release to 99 + 10 pg/ml (n = 8). In the presence of glibenclamide, the response was significantly (p < 0.003) reduced by 39% (n = 8) (Figure 7(d)). The inhibitory effect of glibenclamide on CGRP release further supports a role of KATP channels in migraine pathophysiology.
Discussion
The main results of the present study are that non-selective inhibitors of KATP channels have a significant effect in two validated animal models of migraine, the spontaneous trigeminal allodynic (STA) rat and mice sensitized by repeated injections of GTN. In the rat model, glibenclamide inhibited a chronic state of cephalic hypersensitivity while in the mouse model the 2 h post-drug effect of GTN was prevented whereas basal hyperalgesia progressed. In addition, the KATP channel opener levcromakalim induced hypersensitivity similar to that induced by GTN. Ex vivo studies showed that glibenclamide inhibits CGRP release from dura mater and TG. Our data suggest that KATP channel inhibition is a promising new target for migraine drugs.
Role of blood glucose levels for pain responses
Decreased levels of blood glucose after insulin administration were recently shown to attenuate dural-evoked neuronal firing (45). In our studies with glibenclamide, blood glucose was decreased by 55% and it could therefore be expected to have an impact on the measured mechanical thresholds. However, in experiments performed on wild type SD rats we saw no decrease in sensitivity after glibenclamide administration (Figure 2(b)). The mechanical thresholds in our study were measured at 2 h after glibenclamide administration. In the study by Martins-Oliveira (45), blood glucose levels were shown to be stably decreased at 1 h after insulin treatment. However, at 30 min after the treatment, the dural-evoked firing had already started to decline, which might be indicating the initiation of unknown adapting mechanisms. It could be speculated that the lack of an effect on mechanical thresholds in spite of decreased blood glucose levels is due to adaptive mechanisms being fully activated 2 h after administration of glibenclamide. A key point is provided in Figure 3, where it is shown that 10 and 100 mg/kg of gliquidone lowered blood glucose to the same level. However, only the 100 mg/kg dose significantly increased withdrawal responses in STA rats. Accordingly, we believe that the anti-allodynic effect observed is most likely to be mediated via KATP channel mediated effects within the trigeminovascular system rather than simply via lowering of blood glucose.
KATP channel activation causes opposite pain effects in the periphery compared to the brain
Patch clamp recordings made from cultured nociceptive neuronal preparations exemplify the role of K+ channels within neuronal pain circuits, wherein they act to dampen cellular excitability. Accordingly, the Kv7 channel opener retigabine and the KATP channel opener pinacidil hyperpolarize the somatic membrane potential of cultured dorsal root ganglion neurons (46). Local injection of these substances into the dorsal root ganglion diminishes hindpaw inflammatory hyperalgesia in rats produced by bradykinin (46). A further extensive literature indicates that intracerebroventricular administration of K+ channel openers mediates robust antinociception in an array of inflammatory, post-operative and neuropathic pain models (47). These data are at odds with the current data in both human and rodent models, where levcromakalim induced migraine and hyperalgesia, respectively.
So how can we explain their apparent paradoxical effects on trigeminal nociceptive processing? At this point it is impossible to distill the mechanisms evoked by levcromakalim and glibenclamide as being mediated by only one cell type. The downstream consequences mediated via these channels in, for example, vascular cells versus neurons, is ultimately different. This is a network and ultimately it is the balance of excitation versus inhibition that is measured as glibenclamide efficacy in the animal models. This probably also explains the differences between the trigeminal and spinal pain processing systems in relation to KATP channels. KATP channels are widely expressed within the brain, which theoretically could be a target site (48,49). However, there is no evidence that either levcromakalim 50 or glibenclamide cross the blood-brain barrier. Rats implanted subcutaneously with 200 mg glibenclamide pellets (dose ∼ 50 mg/kg/day) attain a mean plasma concentration in the low micromolar range within 7–10 days, whereas levels within the brain and CSF were undetectable (51). A concentration in the micromolar range is easily sufficient to block inwardly rectifying K+ currents mediated via KATP channels comprised of Kir6.2 and SUR1 subunits located in peripheral tissues (43,44). Moreover, even when given as a bolus injection (50 mg/kg, i.p.), similar to the administration method used here, CSF levels are 1000-fold lower than in plasma (34 µg/ml) (51). Thus, it is likely that drugs such as glibenclamide act at peripherally localized KATP channels to diminish migraine-like pain behaviors of the cephalic pain pathways.
Vascular effects of KATP channel agents
Using a closed cranial window model, we have previously reported that glibenclamide attenuates dural dilatation induced by CGRP and transcranial electrical stimulation in anesthetized rats (52). This is consistent with the extensive localization of KATP channels throughout vascular tissues (53), wherein channel opening within smooth muscle results in membrane hyperpolarization and closure of voltage-operated Ca2+channels. Accordingly, the associated relaxation induces a vasodilation, which we believe can be a primary functional mechanism that promotes hyperalgesia within the trigeminovascular system. It is also consistent with human studies showing extracerebral but no cerebral vasodilation after levcromakalim infusion (50). Crucially, this mechanism appears to be variously conserved for all vasodilatory molecules irrespective of the target and/or signal transduction pathway(s) they engage.
Cellular mechanisms of action in relation to clinical effects
When administered to humans, a single dose of levcromakalim was 100% effective in migraine provocation. In contrast, in mice hypersensitivity was not observed until the third or fifth day. How can this be explained? The studies are performed in two different species and with different routes of administration. Furthermore, the endpoint values are not the same. In the human study, patients report on their headache intensity, while in mice, we investigate changes in mechanical sensitivity. Thus, the results in mice cannot be directly compared to human experiments. The time of onset of the increased sensitivity is different in mice treated with levcromakalim versus GTN. However, GTN releases the gaseous transmitter NO that rapidly diffuses into the cells to activate guanylyl cyclase and forms oxidative radicals, while levcromakalim has to diffuse to its site of action.
It is also interesting that KATP channel opening in man is more effective in provoking migraine than other provoking agents and likewise KATP channel inhibition was very effective in the animal models used in the present study. What are the possible cellular and molecular mechanisms? We hypothesize that the least effective provoking agents act on receptors in the cellular membrane (Figure 8); next come agents like NO, sildenafil and cilostazol that directly affect intracellular second messenger concentrations and, as the most efficient, comes the KATP channel opener levcromakalim. In the cascade of events of at least some pathways, KATP channels are the final common mechanism (54).

KATP channels as the common denominator target for migraine provoking agents. Schematic representation of membrane bound and intracellular targets for migraine provoking agents. Glyceryl trinitrate (GTN) releases nitric oxide (NO), which activates soluble guanylyl cyclase (sGC) catalysing the formation of cyclic guanosine monophosphate (cGMP) that binds to protein kinase G (PKG) causing release of active catalytic subunits of PKG leading to phosphorylation and opening of KATP channels. This system is also affected by the phosphodiesterase 5 (not shown in the figure) inhibitor sildenafil, resulting in accumulation of cGMP. Binding of the migraine provokers calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase activating peptide (PACAP) to their receptors leads to activation of soluble adenylyl cyclase (sAC). This causes increased formation of cyclic adenosine monophosphate (cAMP) that activates protein kinase A (PKA) followed by phosphorylation of KATP channels. The migraine provoker cilostazol increases accumulation of cAMP by inhibition of phosphodiesterase 3 (not shown). The KATP channel is directly activated by levcromakalim and inhibited by glibenclamide.
KATP channel inhibition as a possible new treatment for migraine
Given that KATP channel inhibition may be effective in spontaneous migraine, it is worth speculating about the possibilities of propagating this target. As discussed above, there are several isoforms of the KATP channel. Glibenclamide and gliquidone are relatively unspecific and inhibit most if not all isoforms of the channel. However, gliquidone has a lower selectivity for cardiomyocytes and vascular smooth muscle cells than glibenclamide (33). This might be why gliquidone was less effective in the present study. Both glibenclamide and gliquidone have potent effects on pancreatic β-cells which are associated to SUR1 and induced hypoglycemia makes them unsuitable for migraine therapy (55). Other isoforms such as SUR2A may have cardiac side effects that should preferably be avoided (56). In previous studies, we have analyzed the presence of KATP channel isoforms in the cephalic vasculature and came to the result that inhibitors of the SUR2B channel subunit most likely would be preferred (42,53,57–59). Unfortunately, no such compound with suitable in vivo pharmacokinetic properties was available for testing in the present study. Subtype specific KATP channel inhibitors, preferably targeting SUR2B/Kir6.1, should be developed and tested in our models and in other migraine models before clinical trials can be initiated.
Key findings
KATP channel inhibitors glibenclamide and gliquidone attenuate periorbital hypersensitivity in STA rats. KATP channel opener levcromakalim induces hypersensitivity in mice. Glibenclamide blocks GTN and levcromakalim induced hypersensitivity in mice. Glibenclamide inhibits CGRP release from isolated dura mater and trigeminal ganglion. Inhibition of KATP channels is suggested to be a promising target for development of new migraine drugs.
Footnotes
Author contributions
GM, DMK and SLC designed experiments with advice and guidance from JO and IJ-O. AS, SP, GM and SLC performed the experiments and analysed the data. All authors were involved in the interpretation of the data. GM, JO and IJ-O wrote the paper with contributions from all the other authors.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Candys Foundation.