
Abstract
Background
Calcitonin gene-related peptide (CGRP) is a neuropeptide that acts in the trigeminovascular system and is believed to play an important role in migraine. CGRP activates two receptors that are both present in the trigeminovascular system; the CGRP receptor and the amylin 1 (AMY1) receptor. CGRP receptor antagonists, including olcegepant (BIBN4096BS) and telcagepant (MK-0974), can treat migraine. This study aimed to determine the effectiveness of these antagonists at blocking CGRP receptor signalling in trigeminal ganglia (TG) neurons and transfected CGRP and AMY1 receptors in Cos7 cells, to better understand their mechanism of action.
Methods
CGRP stimulation of four intracellular signalling molecules relevant to pain (cAMP, CREB, p38 and ERK) were examined in rat TG neurons and compared to transfected CGRP and AMY1 receptors in Cos7 cells.
Results
In TG neurons, olcegepant displayed signal-specific differences in antagonism of CGRP responses. This effect was also evident in transfected Cos7 cells, where olcegepant blocked CREB phosphorylation more potently than expected at the AMY1 receptor, suggesting that the affinity of this antagonist can be dependent on the signalling pathway activated.
Conclusions
CGRP receptor antagonist activity appears to be assay-dependent. Thus, these molecules may not be as selective for the CGRP receptor as commonly reported.
Introduction
The precise pathophysiology of migraine is unclear, but is hypothesised to involve dysfunction of the trigeminovascular system. This includes the cranial vasculature, the trigeminal ganglia (TG) and the spinal trigeminal nuclei of the brainstem. The TG contains the cell bodies of the sensory trigeminal neurons that transmit signals from the periphery to the brainstem. Calcitonin gene-related peptide (CGRP), a 37 amino acid neuropeptide, is believed to play an important role in migraine. For example, during a migraine attack CGRP levels are increased in the blood and infusion of CGRP into migraine sufferers can induce a migraine-like attack (1,2). This has resulted in CGRP receiving much clinical attention and in the development of several new investigational drugs against the CGRP system (3,4).
CGRP potently activates two receptors; the CGRP receptor and the AMY1 receptor, a subtype of amylin receptor that is responsive to both CGRP and the related amylin peptide. The two CGRP receptors are formed by the co-expression of receptor activity-modifying protein 1 (RAMP1) with two different G protein-coupled receptors. The CGRP receptor is formed by RAMP1 and the calcitonin receptor-like receptor (CLR), whereas the AMY1 receptor is formed by RAMP1 and the calcitonin receptor (CTR) (5–7). In the trigeminovascular system, both CGRP and AMY1 receptors have been identified (8–11). However, the majority of research and clinical development has focused upon the CGRP receptor. For example, olcegepant (BIBN4096BS) and telcagepant (MK-0974) are small molecule antagonists that bind to the CGRP receptor and block its activation by CGRP (12). Olcegepant and telcagepant have both displayed efficacy in clinical trials but have been discontinued (13,14). These antagonists are generally considered to be CGRP receptor-specific, however both olcegepant and telcagepant can also act as antagonists at the AMY1 receptor (8,15). The potential biological role of the AMY1 receptor in CGRP biology and migraine pathogenesis is not yet clear.
Activation of receptors by CGRP results in the induction of a diverse range of downstream signalling pathways that mediate physiological and pathophysiological effects (16). For example, limited studies in TG and dorsal root ganglia (DRG) neurons have implicated several signalling pathways, including cAMP, cAMP response element-binding protein (CREB), p38 and extracellular signal-regulated kinase (ERK) in CGRP biology (5,17–20). The importance of these pathways in CGRP action or the specific receptor mediating these effects remain unclear.
For pain-relieving drugs, such as morphine, specific signalling molecules are hypothesised to regulate distinct biological effects, including pain relief, tolerance or respiratory dysfunction (21,22). This has led to attempts to design drugs that display signalling bias: drugs that preferentially produce signalling associated with pain relief over signalling associated with side-effects (21,23). Given these developments, a deeper understanding of CGRP signalling and the sensitivity of different pathways to CGRP receptor antagonists in neurons is needed. Therefore, in this study we have profiled the ability of CGRP to activate different signalling pathways and examined the sensitivity of these signalling pathways to CGRP receptor blockade in rat TG neurons and compared the outcomes to defined CGRP and AMY1 receptors in transfected Cos7 cells.
Materials and methods
Isolation and culture of TG neurons
Isolation and culture of TG neurons was performed as described (8,24). Briefly, 4–5-day-old postnatal Wistar rat pups were euthanised by decapitation and the TG collected. This model was used because it is an established model for studying trigeminal neuron signalling. TGs were dissociated by incubation with dispase II (10 mg/mL) for 30 min at 37℃. Cultures were enriched for TG neurons by differential centrifugation through a bovine serum albumin gradient. The TG neuron-enriched pellet was re-suspended in TG neuron culture media (L15 medium containing penicillin/streptomycin, glutamine [2 mM], glucose [50 mM], ascorbic acid [250 µM], glutathione [8 µM], mouse 2.5 S nerve growth factor [NGF; Alomone Labs, Jerusalem, Israel] [10 ng/mL] and 8% fetal bovine serum [FBS]) and further enriched by pre-plating for 1 h at 37℃. TG neurons were then plated (approximately four wells/ganglia) into 384 well poly-D-lysine/laminin-coated (BD Biosciences, New Zealand) cell culture plates. Cultures were maintained at 37℃ in a humidified incubator for 24 h prior to conducting signalling assays. All procedures involving the use of animals at the University of Auckland were conducted in accordance with the New Zealand animal welfare act (1999) and approved by the University of Auckland Animal Ethics Committee.
Phosphorylated ERK histology in TG cultures from Sprague Dawley rats
Glass coverslips were pre-coated with laminin (Gibco, Grand Island, NY, USA) for 1 h at 37℃. Postnatal day 4–5 Sprague Dawley (SD) rats were euthanised by CO2 asphyxiation and TGs were removed and prepared according to procedures outlined above. Following differential centrifugation, neurons were plated at a concentration of two pups per coverslip in L15 media containing 10% FBS, penicillin/streptomycin, amphotericin B, 50 mM glucose, 250 µM ascorbic acid, 8 µM glutathione and 10 ng/mL NGF (Alomone Labs, Jerusalem, Israel). Cells were serum starved for 10 min, then treated with 100 nM rat αCGRP (Sigma-Aldrich, St. Louis, MO, USA) or 20% FBS (Invitrogen, Eugene, OR, USA) for 7 min. Cells were washed and then fixed with 4% paraformaldehyde for 15 min. Cells were permeabilised using 0.3% Triton X-100. Primary antibodies were raised against phospho-p44/42 MAPK (ERK1/2) raised in mouse (#9101L, Cell Signaling Danvers, MA, USA) and neuron-specific beta III tubulin raised in rabbit (#ab78078, Abcam, Cambridge, MA, USA) and used at 1:200 and 1:300, respectively. Secondary antibodies were anti-mouse or anti-rabbit raised in goat, conjugated to Alexa 488 or Alexa 568 and used at 1:500 (Invitrogen, Eugene, OR, USA). Coverslips were mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Images were captured using a Zeiss LSM 710 confocal microscope. All images were captured as z stacks under identical exposure conditions. Image J was used for image analysis. Briefly, circular fields surrounding neuron cell bodies were selected and mean grey value in the pERK1/2 channel was measured. The total number of fields measured was 51 neurons from ten random fields for vehicle, 58 neurons from eight random fields for 100 nM rαCGRP and 53 neurons measured from 11 random fields for 20% FBS. Animal care and procedures were approved by the University of Iowa Animal Care and Use Committee and performed in accordance with the standards set by the National Institutes of Health.
Cell culture and transfection
Culture of Cos7 cells was performed as previously described (6,8). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 8% heat inactivated FBS and kept in a 37℃ humidified 5% CO2 incubator. Cells were seeded into 96-well plates at a density of 15,000–20,000 cells per well (determined using a Countess Counter™, Invitrogen) one day prior to transfection. Cells were transiently transfected using polyethylenimine as described previously (6,8). Hemagglutinin-tagged human CLR, hemagglutinin-tagged human CTR (CT(a) subtype) and myc-tagged human RAMP1 in pcDNA3 or 3.1 plasmid vectors were under the control of the human cytomegalovirus promotor and used as previously described (6,7).
Signalling assays in rat TG neurons and transfected cells
cAMP assays in TG neurons were performed as previously described (24). Briefly, assays in TG neurons were performed in serum and NGF-free media containing 0.1% BSA. TG neurons were serum starved for 5 min, then incubated with media alone or 10 pM to 10 µM rat αCGRP with or without antagonists for 30 min at room temperature. cAMP assays were performed with 1 mmol/L 3-isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich, St.Louis, MO, USA). cAMP content was determined using the LANCE ultra cAMP detection kit (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA). cAMP was measured in transfected Cos7 cells as described previously with minor modifications for use of the LANCE cAMP detection kit (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA) (6). These cAMP assays were also performed with 1 mM IBMX (Sigma-Aldrich, St. Louis, MO, USA). Following incubation with media alone or 1 pM to 1 µM human αCGRP, human amylin or rat amylin in the presence or absence of antagonist for 15 min at 37℃ the contents of the wells were aspirated and the reaction stopped by the addition of 50 µL absolute ethanol. cAMP samples were then stored at –30℃ for up to seven days before further analysis. Absolute ethanol was evaporated off the cAMP samples in a fume hood and cAMP extracted from dry samples by addition of 40 µL cAMP detection buffer (0.35% Triton X-100, 50 mM HEPES and 10 mM calcium chloride). Samples were then gently shaken at room temperature for 10–15 min. Five microlitres of cAMP sample was then transferred to a white 384 well optiplate and cAMP measured using the LANCE cAMP detection kit (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA) as per the manufacturer’s instructions. The concentration of cAMP in each sample was determined from a standard curve that was generated in parallel for both LANCE and LANCE ultra assays.
Phosphorylated ERK1/2 (pERK1/2), pCREB and p38 (pp38) were measured using AlphaScreen Surefire or AlphaLisa Surefire ULTRA assay kits (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA). Surefire assays are a well-characterised and sensitive method for the detection of phosphoproteins. These assays display equivalent or greater sensitivity than western blotting and other methodologies (25–27). Preliminary time-course experiments were conducted to determine the optimal time to measure signalling (data not shown). Following peptide stimulation at 37℃ for 7 min (ERK1/2) or 30 min (CREB and p38), media was removed using an eight-channel aspirator (Cos7 cells) or a 27-gauge syringe (neurons). Twenty microlitres (Cos7) or 6 µL (neurons) of lysis buffer was added to each well and the plates incubated with gentle shaking at room temperature for 10–15 min. Four microlitres of each sample were transferred into a white 384-well optiplate and Surefire assays performed as per the manufacturer’s instructions.
All assay plates were read using an Envision plate reader (Perkin-Elmer). Human (h) and rat (r) αCGRP were purchased from American Peptide (Sunnyvale, CA, USA) or Bachem (Bubendorf, Switzerland). rAmylin, rαCGRP8-37 and hαCGRP8-37 were purchased from American Peptide (Sunnyvale, CA, USA). hAmylin was synthesised in-house by Fmoc Solid-phase peptide synthesis (28). Peptide content was assumed to be 80% when not known. Olcegepant was kindly provided by Boehringer-Ingelheim (Ingelheim, Germany). Telcagepant was purchased from Suzhou Rovathin Pharmatech Co. (Jiangsu, China). Forskolin (Tocris, Bristol, UK) or FBS were used as positive controls in cAMP and CREB or p38 and ERK1/2 assays, respectively.
Data analysis
All statistical analysis and curve fitting were performed using Graphpad Prism 6.0 (GraphPad Software, San Diego, CA, USA). Maximal signalling responses (Emax) were determined and the data are expressed as a percentage of the Emax for the associated control curves to enable combined data to be presented in figures. For pERK1/2 assays in TG neurons the data are expressed as a percentage of 20% FBS. For p38 assays in Cos7 cells, data were normalised to the media only control (basal) and presented as fold basal in concentration-response curve experiments. To define agonist potency, pEC50 values were obtained by fitting a four-parameter logistic equation to the concentration-response curve data. F-tests were then performed to determine if the Hill slope was significantly different from one. For the majority of individual experiments, the Hill slope was not significantly different to unity. Agonist potency curves were therefore re-fitted with a Hill slope constrained to one to obtain pEC50 values for all experiments. To define antagonist potency, pA2 values were calculated from pEC50 values obtained in the presence or absence of antagonist. F-tests were performed within each individual dataset to determine whether the presence of antagonist significantly shifted the concentration response curve. When the majority of individual experiments reported a significant difference, pA2 values were calculated. For statistical analysis, pEC50 and pA2 values from individual experiments were combined and significant differences determined using two-tailed Student’s t-tests or one-way ANOVA with post hoc Dunnett’s tests. Statistical significance was defined as p < 0.05. All data points represent the mean ± SEM combined from n separate experiments. Separate experiments comprise individual TG neuron or transiently transfected Cos7 preparations, performed with triplicate wells in each experiment.
Results
CGRP stimulation and olcegepant antagonism of cAMP, CREB, p38 and ERK1/2 signalling in TG neurons
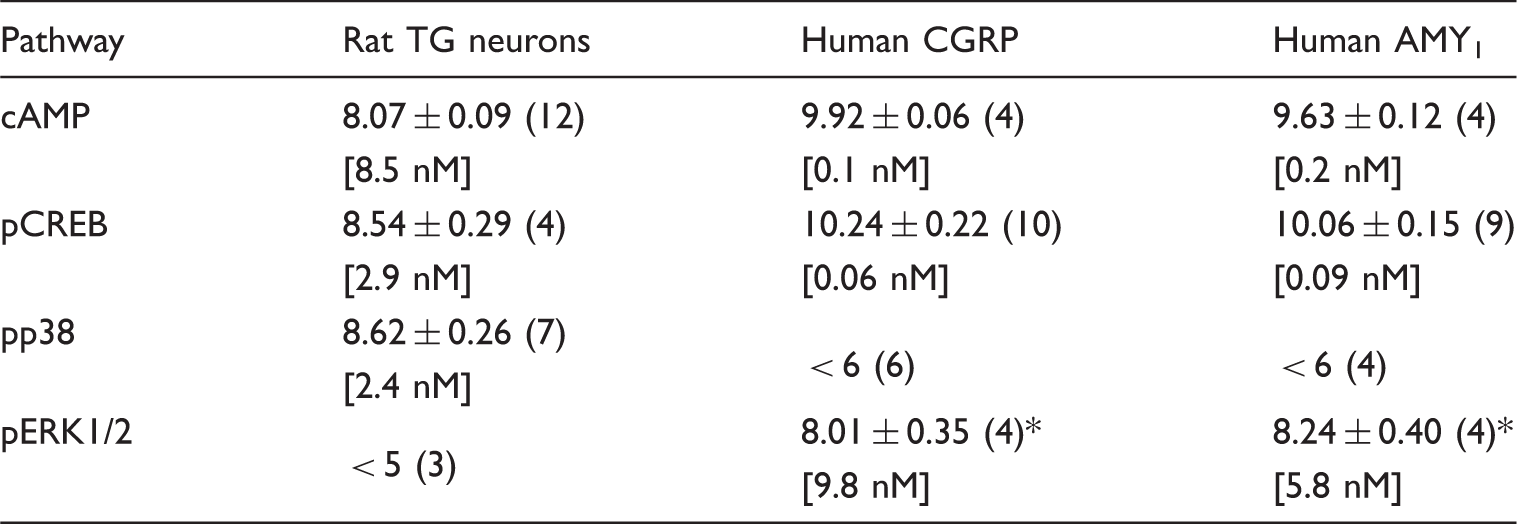
In TG neurons, rαCGRP increased CREB phosphorylation (Figure 1a) and p38 phosphorylation (Figure 1b) with potencies that were not significantly different to those observed previously for cAMP (8) (Table 1). Interestingly, 10 µM rαCGRP consistently produced greater CREB phosphorylation than 1 µM, suggesting that CREB activation is biphasic and may involve multiple mechanisms. rαCGRP did not induce ERK1/2 phosphorylation in either acute time course (0–30 min using 100 nM rαCGRP; data not shown) or concentration response studies (Figure 1c). To ensure that the methods used had not failed to detect phosphorylated ERK1/2, the ability of αCGRP to cause ERK1/2 phosphorylation was measured using a distinct histological method. This confirmed a lack of detectable acute ERK1/2 phosphorylation in response to αCGRP in neurons and other cell types present under these conditions (Figure 1d; quantification not shown). Therefore, ERK1/2 phosphorylation was not measured in further TG neuron experiments.
αCGRP-stimulated CREB, p38 and ERK phosphorylation in rat trigeminal ganglia neurons. Stimulation of (a) CREB, (b) p38 and (c) ERK1/2 phosphorylation by αCGRP and 20% FBS (inset), **p < 0.01. (d) Histological detection of ERK1/2 phosphorylation in vehicle, 100 nM αCGRP and 20% FBS treated cultures. Data points are mean ± SEM of 3–7 combined experiments (a–c) or three independent preparations (d). Summary of CGRP potencies (pEC50) for cAMP accumulation, CREB, p38 or ERK1/2 phosphorylation in neuron-enriched rat TG cultures and Cos7 cells transfected with human CGRP receptor components. Data are mean ± SEM of n individual experiments in parentheses. Rat TG neuron cAMP data are from Walker et al., 2015 (8). Comparisons were performed by one-way ANOVA followed by post-hoc multiple comparisons tests. p < 0.05 vs. cAMP and pCREB.
To better define how olcegepant blocks CGRP activity in TG neurons, the ability of olcegepant to block activation of CREB and p38 was determined. Olcegepant effectively antagonised rαCGRP-induced CREB phosphorylation and p38 phosphorylation (Figure 2, Table 2). p38 phosphorylation was antagonised with similar potency to that observed previously for cAMP accumulation (8). Surprisingly, antagonism of CREB phosphorylation by olcegepant was approximately 20-fold greater than that observed for cAMP (Table 2).
Olcegepant blocks αCGRP-stimulated CREB and p38 phosphorylation in rat trigeminal ganglia neurons. Antagonism of αCGRP-stimulated (a) CREB and (b) p38 phosphorylation by 100 nM olcegepant. Data points are mean ± SEM of four or five combined experiments. Summary of the ability of olcegepant and telcagepant to antagonise (pA2) αCGRP-mediated cAMP accumulation and CREB or p38 phosphorylation in neuron-enriched rat TG cultures or Cos7 cells transfected with human CGRP or AMY1 receptors. Data are mean ± SEM of n individual experiments in parentheses. Rat TG neuron cAMP data are from Walker et al., 2015. Comparisons were performed by one-way ANOVA followed by post-hoc multiple comparisons tests or Student’s t-tests. p < 0.05 vs. cAMP. N.D. = not done.

To confirm that CGRP signalling was attributable to TG neurons, we also conducted experiments in TG cultures enriched for non-neuronal glial cells. rαCGRP (100 nM) did not stimulate cAMP accumulation or induce ERK1/2, p38 and CREB phosphorylation (Supplemental Figure 1). However, rαCGRP potentiated cAMP accumulation in the presence of 50 µM forskolin, suggesting that these cells have a weak capacity to respond to CGRP.
CGRP stimulation of cAMP, CREB, p38 and ERK signalling at CGRP and AMY1 receptors in transfected Cos7 cells
In a transfected Cos7 cell model, hαCGRP increased cAMP accumulation (Figure 3a) and CREB phosphorylation (Figure 3b) at both CGRP and AMY1 receptors. The activity of hαCGRP at both these receptors was equipotent (Table 1). Notably, 1 µM hαCGRP consistently produced greater CREB phosphorylation than 100 nM at the AMY1 receptor. This was occasionally observed at the CGRP receptor. Interestingly, no p38 phosphorylation was observed in response to hαCGRP in transfected Cos7 cells (Figure 3c, Table 1). This suggests that p38 is not coupled to either CGRP or AMY1 receptor activation in this Cos7 cell model. Despite the lack of rαCGRP-induced ERK1/2 phosphorylation in TG neurons, ERK1/2 phosphorylation was measurable in transfected Cos7 cells. In the Cos7 cellular background hαCGRP increased ERK1/2 phosphorylation (Figure 3d) at both CGRP and AMY1 receptors with similar potency. ERK1/2 activation was, however, approximately 100-fold weaker than cAMP or CREB activation at CGRP and AMY1 receptors (Table 1). These results indicate that cellular background may play an important role in αCGRP-mediated p38 and ERK1/2 activation. Further investigation of p38 or ERK1/2 activation was not conducted in the Cos7 cellular background.
αCGRP-stimulated cAMP accumulation and CREB, p38 and ERK phosphorylation in Cos7 cells transfected with CGRP and AMY1 receptors. Stimulation of (a) cAMP accumulation and (b) CREB, (c) p38 and (d) ERK1/2 phosphorylation by αCGRP. Data points are mean ± SEM of 3–10 combined experiments.
Effect of olcegepant on CGRP-stimulated cAMP and CREB signalling at CGRP and AMY1 receptors in transfected Cos7 cells
Olcegepant antagonised hαCGRP-stimulated cAMP accumulation at hCGRP and hAMY1 receptors (Figure 4a and 4b). Olcegepant was approximately 130-fold more potent at the hCGRP receptor than the hAMY1 receptor (Figure 4c, Table 2). Olcegepant antagonised αCGRP-stimulated CREB phosphorylation with similar potency to cAMP accumulation at hCGRP receptors (Figure 4d, Table 2). However, mirroring the results observed in TG neurons, at the hAMY1 receptor αCGRP-stimulated CREB phosphorylation was antagonised significantly more potently than cAMP accumulation (Figure 4e). Olcegepant was only 26-fold more potent at hCGRP receptors than hAMY1 receptors (Figure 4f, Table 2). This suggests that blockade of the AMY1 receptor is responsible for the more potent antagonism of CREB phosphorylation observed for olcegepant in TG neurons. ERK1/2 phosphorylation by αCGRP was too weak to produce meaningful antagonist measurements in transfected Cos7 cells.
Olcegepant blocks αCGRP-stimulated cAMP accumulation and CREB phosphorylation in Cos7 cells transfected with CGRP and AMY1 receptors. Antagonism of αCGRP-stimulated cAMP accumulation at (a) CGRP and (b) AMY1 receptors by olcegepant. (c) Comparison of olcegepant antagonism of αCGRP-stimulated cAMP accumulation at CGRP and AMY1 receptors. Antagonism of αCGRP-stimulated CREB phosphorylation at (d) CGRP and (e) AMY1 receptors by olcegepant. (f) Comparison of olcegepant antagonism of αCGRP-stimulated CREB phosphorylation at CGRP and AMY1. Data points are mean ± SEM of 5–9 combined experiments. ***p < 0.001.
Effect of olcegepant on amylin-stimulated cAMP signalling at CGRP and AMY1 receptors in transfected Cos7 cells
To better define the activity of olcegepant at AMY1 receptors, the ability of olcegepant to block amylin-induced cAMP accumulation was determined (Figure 5). Olcegepant blocked human and rat amylin with pA2 values of 6.72 ± 0.13 (n = 4) and 6.70 ± 0.20 (n = 3), respectively. Surprisingly, olcegepant was approximately 14-fold more potent at blocking αCGRP than human or rat amylin at AMY1 receptors (see Table 2 for CGRP data). This suggests that, compared with αCGRP, amylin is relatively resistant to blockade by olcegepant in AMY1 receptor transfected Cos7 cells and indicates that direct comparisons between αCGRP and amylin signalling behaviours is not straightforward. Experiments with amylin at the CGRP receptor were not performed because amylin is a very weak agonist at this receptor (6,29).
Olcegepant blocks human (h) amylin and rat (r) amylin-stimulated cAMP accumulation in Cos7 cells transfected with AMY1 receptors. Antagonism of (a) hAmylin and (b) rAmylin-stimulated cAMP accumulation at AMY1 receptors by 1 µM olcegepant. Data points are mean ± SEM of three or four combined experiments.
Effect of telcagepant and CGRP8-37 on CGRP-stimulated cAMP and CREB signalling at CGRP and AMY1 receptors in transfected Cos7 cells
To determine if the more potent antagonism of CREB phosphorylation at the AMY1 receptor was specific to olcegepant, experiments were repeated with telcagepant. In line with our previous publications, telcagepant antagonised αCGRP-stimulated cAMP accumulation at hCGRP and hAMY1 receptors (Figure 6a and 6b). Telcagepant was approximately 35-fold more potent at hCGRP receptors than hAMY1 receptors (Figure 6c, Table 2). Antagonism of αCGRP-stimulated CREB phosphorylation by telcagepant was not significantly different to that observed for cAMP accumulation at either hCGRP or hAMY1 receptors (Figure 6d and 6e), Table 2). Telcagepant blocked CREB phosphorylation approximately tenfold more potently at hCGRP receptors than hAMY1 receptors (Figure 6f, Table 2). This suggests that this mechanism maybe specific to olcegepant; however, as telcagepant is less selective for the hCGRP receptor over the hAMY1 receptor when measuring cAMP accumulation, this effect may be masked (Table 2).
Telcagepant blocks αCGRP-stimulated cAMP accumulation and CREB phosphorylation in Cos7 cells transfected with CGRP and AMY1 receptors. Antagonism of αCGRP-stimulated cAMP accumulation at (a) CGRP and (b) AMY1 receptors by telcagepant. (c) Comparison of telcagepant antagonism of αCGRP-stimulated cAMP accumulation at CGRP and AMY1 receptors. Antagonism of αCGRP-stimulated CREB phosphorylation at (d) CGRP and (e) AMY1 receptors by telcagepant. (f) Comparison of telcagepant antagonism of αCGRP-stimulated CREB phosphorylation at CGRP and AMY1. Data points are mean ± SEM of five combined experiments. *p < 0.05, ***p < 0.001.
CGRP8-37 is another commonly used antagonist of CGRP responses, although it is not a good discriminator of CGRP and AMY1 receptors (8). Experiments conducted examining cAMP responses showed that human and rat CGRP8-37 displayed no measurable agonist activity at CGRP receptors (Figure 7a). However, at AMY1 receptors, both human and rat CGRP8-37 displayed weak partial agonist activity (Figure 7b). At the AMY1 receptor, human CGRP8-37-stimulated cAMP with a pEC50 of 5.87 ± 0.19 (n = 4) and an Emax of 44.17% ± 9.37 (n = 4) compared to αCGRP. Similarly, rat CGRP8-37 gave a pEC50 of 7.49 ± 0.22 (n = 4) and an Emax of 19.45% ± 2.89 (n = 4). This partial agonism stimulated by CGRP8-37 complicates direct comparisons of antagonism at CGRP and AMY1 receptors and precluded further analysis.
Stimulation of cAMP accumulation by αCGRP, human (h) CGRP8-37 and rat (r) CGRP8-37 in Cos7 cells transfected with CGRP and AMY1 receptors. (a) CGRP and (b) AMY1 receptors. Data points are mean ± SEM of four combined experiments.
Discussion
Several antagonists of the CGRP receptor, called the ‘gepants’, have been developed and entered into clinical trials for the treatment of migraine (4). These antagonists have shown great promise. However, currently no gepant has reached the market and several questions regarding efficacy have been raised, leading to the suggestion that other factors may be involved in migraine (30,31). We recently showed that the AMY1 receptor, a second CGRP receptor, was expressed in TG and proposed that this receptor may be important for CGRP activity in migraine (8). Closer examination of the ability of αCGRP to activate signalling in relevant neurons or at defined CGRP and AMY1 receptors is required to exploit the CGRP system to its fullest potential. Herein, we report the ability of CGRP to stimulate cAMP production, p38, ERK1/2 and CREB phosphorylation and the signalling molecule-dependent ability of olcegepant to block CGRP action in TG neurons and transfected cells.
The binding of CGRP to its receptors is reported to facilitate conformational changes in the receptor that lead to the activation of diverse downstream signalling pathways (16,32). The major downstream signalling event described for the activation of both CGRP and AMY1 receptors is the accumulation of cAMP (6–8). We have built on these studies to generate a more complete picture of the pharmacology involved in CGRP-meditated signalling and assess the potential different pathways that could be activated. The activation of the mitogen-activated protein kinase, ERK, has commonly been used as a marker of neuron activation; however, CREB phosphorylation may be appropriate for specific biological functions (18,33,34).
We observed potent αCGRP-stimulated CREB phosphorylation in TG neurons. Similarly, high potency was obtained at CGRP and AMY1 receptors in Cos7 cells. Although the stimulation of CREB phosphorylation by αCGRP has previously been reported in cultured TG neurons, the potencies of αCGRP and olcegepant have never been quantified for this important signalling molecule (18). Unexpectedly, olcegepant displayed significantly greater antagonism of αCGRP-stimulated CREB phosphorylation compared to cAMP accumulation in TG neurons. Although agonist signalling bias is becoming increasingly common, with reports including both synthetic and natural ligands (22,24,35), signalling molecule-dependent antagonist bias has rarely been reported and is not well described or understood (36). Given this observation, coupled with the potentially pivotal role CREB plays in biological functions including morphine tolerance and drug dependence/addiction (33,34,37,38), we examined the ability of olcegepant to block CREB activation at CGRP and AMY1 receptors. Olcegepant displayed significantly greater antagonism of αCGRP-stimulated CREB phosphorylation than cAMP accumulation at the AMY1 receptor, but not at the CGRP receptor in transfected Cos7 cells. This suggests that olcegepant may be more active at AMY1 receptors than is currently appreciated and that the precise signalling mechanisms resulting in CREB activation may differ between CGRP and AMY1 receptors. Additional experiments examining telcagepant activity in transfected cells suggest that this property maybe unique to olcegepant. However, there was a trend towards reduced selectivity between CGRP receptors for telcagepant when measuring pCREB, suggesting that this effect could extend to other molecules but this would need to be tested in future studies.
The activity of αCGRP and anti-migraine drugs at the AMY1 receptor should also be considered in regard to potential side-effects, e.g. higher than expected blockade of αCGRP could have potential cardiovascular effects (39). Given the important role of amylin and amylin receptors in controlling food intake and obesity (40), chronic blockade of the AMY1 receptor could have side-effects on metabolism. In the current study, olcegepant displayed relatively low antagonism of amylin compared to αCGRP in AMY1 receptor transfected Cos7 cells. A similar phenomenon has been observed by others (41). These findings suggest that at the AMY1 receptor olcegepant may display agonist-dependent antagonism. However, cells transfected with AMY1 receptor components are also likely to express amylin-responsive calcitonin receptors which are resistant to olcegepant antagonism (15,42). Although, this finding may be an artefact of the cell culture models required to study these receptors, multiple amylin-responsive receptors may be common in a single cell and therefore representative of typical olcegepant action in vivo. For example, single neurons in the area postrema, which are activated by amylin to control food intake, have been shown to express the components of AMY1, AMY2, AMY3 and calcitonin receptors (43). It is not known how our observations would affect other small molecules in development, such as ubrogepant, rimagepant or atogepant (44,45).
Historically, CGRP8-37 has been used as a molecular tool to discriminate between the CGRP receptor and other CGRP-responsive receptors, including amylin and adrenomedullin receptors (46,47). However, CGRP8-37 does not effectively discriminate between CGRP and AMY1 receptors sufficiently (8). This is perhaps unsurprising as both CGRP and AMY1 receptors are effectively activated by CGRP and share a common subunit: RAMP1 (48,49). Interestingly, in the current study CGRP8-37 displayed partial agonism at transfected AMY1 receptors. We observed a weak cAMP response to both rat and human CGRP8-37 at the AMY1 receptor, but no response at the CGRP receptor. It is unclear whether the partial agonism we observed at AMY1 receptors is batch-dependent or cell system-dependent. Interestingly, a similar effect was observed in the first description of CGRP8-37 as an antagonist, where LLC-PK1 cells, which presumably express the calcitonin receptor subunit of an amylin receptor, were weakly activated by CGRP8-37 (50). However, we have not previously observed this level of partial agonism with CGRP8-37, suggesting that there may be variability in CGRP8-37 preparations (7,8). Variable or batch-dependent partial CGRP8-37 agonism may result in apparent resistance to this antagonist, particularly as high concentrations are often required to block a response. Therefore, care should be taken when employing this antagonist. For the most effective use of CGRP8-37, its activity should be assessed in well-characterised cell culture models or used at multiple concentrations, to determine if any partial agonism is present. Where practical, the use of a second antagonist, such as olcegepant or AC187 (7,40), should also be considered.
Recent focus in the CGRP and migraine field has been on the development of antibodies, designed to block CGRP activity (4,45). Anti-CGRP antibodies are currently being designed to block CGRP activity at all potential receptors, including CGRP and AMY1, by preventing CGRP from accessing these receptors. This approach is unlikely to feature signalling pathway-dependent antagonism as the receptors should not interact with the antibodies. However, the sites of expression of each receptor (peripheral and/or central) and thus relative accessibility of the antibodies to the receptors would presumably have an impact on efficacy and/or safety. The second approach, exemplified by AMG334, is to develop anti-CGRP receptor antibodies (4,45). In principal, a CGRP receptor targeted antibody would not block αCGRP activity at that AMY1 receptor. This could result in lower efficacy and/or a reduced side-effect profile. The current evidence does not rule out activity for AMG334 at the AMY1 receptor because the receptor composition of the cells used is undefined (51,52). If AMG334 is selective for the CGRP receptor over the AMY1 receptor, the efficacy of this antibody, coupled with data from the anti-CGRP antibody trials, may shed light onto the role of AMY1 receptors in migraine. However, the efficacy of a drug/monoclonal antibody depends on the dosage regime, half-life and distribution to the site (or sites) of action, which will likely vary between these treatments. Consequently, clinical data for a single drug, such as AMG334, is unlikely to conclusively show the value of targeting the CGRP receptor over the AMY1 receptor.
Traditionally, CREB activation is associated with cAMP accumulation; however, in TG neurons CREB phosphorylation was reportedly dependent on Ca2+/calmodulin-dependent kinase II, which can act independently of cAMP (18,53). Interestingly, the CREB response in TG neurons appeared to be biphasic, indicating the potential presence of a second receptor with significantly lower potency for αCGRP. Given that the biphasic response was also observed at AMY1 receptors, the calcitonin receptor, which can travel to the cell surface independently of RAMPs, is the likely candidate (7).
The stimulation of ERK phosphorylation by CGRP has been observed in several studies involving both non-neuronal and neuronal tissues (54–57). In contrast, we did not observe the acute phosphorylation of ERK1/2 in TG neurons. This difference could be attributed to a lack of direct ERK activation in this cellular background. In other studies, increased ERK phosphorylation could be an indirect effect controlled by secondary factors released by or co-released with CGRP (54,55). Interestingly, a recent study reported that infusion of αCGRP into rats did not alter ERK phosphorylation in the TG, supporting the lack of direct activation (58). We also examined ERK1/2 phosphorylation at CGRP and AMY1 receptors in Cos7 cells. Consistent with previous studies examining amylin and calcitonin activity at AMY1 receptors, in Cos7 cells we observed that αCGRP was considerably weaker at activating ERK1/2, compared to cAMP at both CGRP and AMY1 receptors. This suggests that ERK1/2 activation may occur, at least partly, independently of cAMP accumulation and be activated by G protein-independent mechanisms, such as β-arrestin (16,59). Given that the αCGRP-mediated responses we observed were generally less potent in TG neurons than transfected Cos7 cell models, it is possible that the concentrations of αCGRP used in the current study were simply not high enough to elicit an ERK1/2 response in TG neurons. However, a αCGRP-mediated ERK response at concentrations higher than 10 µM is unlikely to be of physiological relevance. We cannot rule out the possibility that high background ERK phosphorylation in our TG neurons could be masking a response in a small proportion of cells. Interestingly, cellular background appeared to be important for p38 phosphorylation. We observed potent rαCGRP-mediated p38 phosphorylation in TG neurons; however, in the Cos7 cellular background hαCGRP failed to induce a measurable phosphorylation of p38.
An interesting observation in the current study was the apparent lack of CGRP responses from TG glia preparations. CGRP receptor components have been identified histologically in satellite glia (11,60). Studies have also shown that ∼1 µM CGRP can acutely elevate intracellular Ca2+ and chronically induce changes in MAPK signalling and gene transcription in cultured TG glia (61–63). On the other hand, we are only aware of a single study that reports cAMP accumulation in response to CGRP in TG glia (64). Although we observed potent responses to CGRP in TG neurons, 100 nM CGRP did not acutely activate cAMP, ERK1/2, p38 or CREB in our glia-enriched TG cultures. However, 100 nM CGRP amplified forskolin-stimulated cAMP production. This is a well-described phenomenon associated with forskolin co-administration and may indicate low expression of CGRP-responsive receptors in our cellular model (65). This is consistent with previous reports suggesting that TG-derived glia had weaker CGRP receptor staining compared to neurons (66). It is likely that there is variation between TG glia culture models due to the age and sex of animals used, differential regulation of CGRP receptor components due to differences in culture conditions and time in culture (38). Inflammatory mediators may also regulate CGRP receptor component or signalling apparatus expression, leading to relative differences in sensitivity to a CGRP stimulus. Further investigation of CGRP responses in TG glia are warranted in future studies.
Despite the importance of CGRP in migraine, the relative involvement of CGRP and AMY1 receptors in this condition has not been well characterised. It is not known whether activation of either receptor alone is capable of causing a migraine. The presence of the AMY1 receptor at sites important for migraine pain and the side-effects of pain-relieving medications, coupled with the ability of olcegepant to block CREB activation may have consequences for the design and development of new anti-migraine agents. Our observation of signalling pathway-dependent antagonism by olcegepant illustrates some of the complexity in determining drug affinity, and thus selectivity between receptors. In light of our findings, we propose that the role of AMY1 in olcegepant activity may be underappreciated and require deeper investigation.
Article highlights
In trigeminal ganglia neurons and at AMY1 receptors, olcegepant displayed more potent antagonism of CGRP-stimulated CREB phosphorylation than cAMP accumulation. CGRP receptor antagonist activity appears to be pathway-dependent and some molecules may not be as selective for the CGRP receptor as is commonly reported.
Footnotes
Acknowledgements
Olcegepant was kindly provided by Dr. Henri Doods (Boehringer Ingelheim Pharma GmbH & Co., Germany). CSW and DLH conception and design of research; CSW, MJW and ACR performed experiments; CSW analysed data; CSW, AFR and DLH interpreted results of experiments; CSW and ACR prepared figures; CSW and DLH drafted the manuscript; CSW and DLH edited and revised manuscript; CSW, AFR, MJW, ACR and DLH approved the final version of manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Auckland Medical Research Foundation (to CSW and DLH), the University of Auckland Biopharma Thematic Research Initiative (to DLH) and by National Institutes of Health grants R01 NS075599 (to AFR) and F31 NS074728 (to ACR).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.