
Abstract
Background
This study reports the long-term safety and efficacy of erenumab in chronic migraine patients.
Methods
This was a 52-week open-label extension study of a 12-week double-blind treatment phase study. During the double-blind treatment phase, patients received placebo or once-monthly erenumab 70 mg or 140 mg. During the open-label treatment phase, the initial monthly dose was erenumab 70 mg. Following protocol amendment, patients continued to receive erenumab 70 mg if they had already completed their Week 28 visit, otherwise, patients switched from 70 mg to 140 mg; if enrolled after the amendment, patients received 140 mg monthly throughout.
Results
In all, 451/609 (74.1%) enrolled patients completed the study. The exposure-adjusted patient incidence rate for any adverse event was 126.3/100 patient-years for the overall erenumab group. Overall, the adverse event profile was similar to that observed in the double-blind treatment phase. Adverse event incidence rates did not increase with long-term erenumab treatment compared with the double-blind treatment phase, and no new serious or treatment-emergent events were seen.
Efficacy was sustained throughout the 52 weeks. Clinically significant reductions from double-blind treatment phase baseline (about half) were observed for monthly migraine days and migraine-specific medication days. Achievement of ≥50%, ≥75% and 100% reductions from the double-blind treatment phase baseline in monthly migraine days at Week 52 were reported by 59.0%, 33.2% and 8.9% of patients, respectively, for the combined dose group. A numerically greater benefit was observed with 140 mg compared with 70 mg at Weeks 40 and 52.
Conclusions
Sustained efficacy of long-term erenumab treatment in patients with chronic migraine is demonstrated, with safety results consistent with the known safety profile of erenumab and adverse event rates comparable to placebo adverse event rates in the double-blind treatment phase.
Trial registration:
This study is registered at ClinicalTrials.gov (NCT02174861)
Introduction
Chronic migraine (CM) is one of the most prevalent types of headache encountered in tertiary care (1). The high number of migraine days per month in patients with CM impacts patients’ quality of life (2–4). Treatment goals for management of CM include reduction in migraine frequency, reduction in use of acute medications, and safety and tolerability at the administered doses. Conventional oral preventive therapies have been the standard of care for migraine, but are associated with low persistence and adherence rates often leading to frequent switching or complete cessation of treatment (5, 6). As patients with CM often require long-term preventive therapy, an effective and tolerable preventive treatment for CM addresses a major unmet need.
The calcitonin gene-related peptide (CGRP) binds to the canonical CGRP receptor but can also bind to other receptors in the calcitonin-like receptor family including the amylin (to which CGRP binds with comparable affinity as does amylin itself) and adrenomedullin receptors (7). Erenumab, a fully human monoclonal antibody, selectively targets and blocks the canonical CGRP receptor with no affinity for any related receptors (7). The efficacy and safety of erenumab 70 mg and 140 mg has been demonstrated in both episodic migraine and CM (8–11). Erenumab at doses of 70 mg or 140 mg monthly is approved in the United States for the preventive treatment of migraine in adults (12) and in the EU for prophylaxis of migraine in adults who have at least four migraine days per month (13).
The long-term safety and tolerability profile and efficacy of erenumab in patients with episodic migraine are being studied in a 5-year, open-label study, and the 1-year preplanned interim analysis results have been published (14). Here, we report the long-term safety, tolerability and efficacy of erenumab for CM during a 1-year open-label treatment phase (OLTP). The results from the parent 12-week, randomized, placebo-controlled trial were published previously (11).
Methods
Study design
This was a multicenter, 52-week, open-label study following a 12-week double-blind placebo-controlled study, conducted from June 2014 to May 2017 at 64 centers across North America and Europe (NCT02174861). Patients who completed the 12-week parent study and met all eligibility criteria for the OLTP were enrolled within 14 days of completion of the double-blind study. The initial erenumab dose used in the OLTP was 70 mg once monthly. The protocol was subsequently amended to increase the dose to 140 mg monthly to have more long-term safety data from CM patients on the higher dose. Following the protocol amendment, patients who had already completed the Week 28 visit continued to receive erenumab 70 mg monthly for the remainder of the study; patients who had not completed the Week 28 visit increased the open-label erenumab dose from 70 mg to 140 mg monthly at the next visit, and patients who enrolled after the protocol amendment received erenumab 140 mg monthly throughout the study (Figure 1). Concomitant treatments prohibited during the study are listed in Supplemental material, Appendix 1.

Study design.
Eligibility criteria
Patients of either gender, aged 18–65 years with a history of CM (with or without aura) of at least one year, who completed the double-blind study without discontinuing the study treatment and were considered appropriate for continued treatment, were included.
Patients were excluded for the following reasons: Development of an unstable or clinically significant medical condition, laboratory or electrocardiogram abnormality that, in the opinion of the investigator, would pose a risk to patient safety or interfere with study evaluation following randomization into the double-blind study; demonstrated poorly controlled hypertension following randomization into the double-blind study; or systolic blood pressure ≥160 mm Hg and/or diastolic blood pressure ≥100 mm Hg at screening/Day 1. Women of child-bearing potential not on an acceptable and effective mode of contraception, as well as pregnant and nursing women, were also excluded. Eligibility criteria for enrollment in the double-blind study have been reported previously (11).
The final study protocol, informed consent form and accompanying materials provided to study patients were all reviewed and approved by an Independent Ethics Committee or Institutional Review Board at all participating sites. Details of the participating centers are listed in the Supplemental material, Appendix 2. This study was conducted in accordance with International Council for Harmonization Good Clinical Practice regulations/guidelines and in accordance with the ethical principles set forth in the Declaration of Helsinki. Written informed consent was obtained from each patient prior to enrollment.
Outcomes
The primary endpoint was the incidence of adverse events (AEs). The AE grading was based on the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, and AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRa) version 20.0. Secondary endpoints included evaluation of the efficacy parameters; that is, change from DBTP baseline to Week 52 in monthly migraine days (MMD), proportion of patients achieving ≥50% reduction in MMD and change from baseline in monthly acute migraine-specific medication days (MSMD). In order to explore whether there might be a difference in efficacy between the 70 and 140 mg doses, a post-hoc analysis of efficacy parameters was conducted in patients who completed the 52-week OLTP treatment based on the last dose received. Additional post-hoc exploratory endpoints included achievement of ≥75% and 100% reduction in MMD and change from baseline to Week 52 in MSMD in the subset of patients using migraine-specific medications at baseline.
A qualified migraine was defined as a migraine with or without aura lasting at least 4 hours and expressing at least two headache-associated features or at least one associated non-headache feature, or both (Supplemental material, Appendix 2). Any calendar day on which acute migraine-specific medication (triptan in 99% of the cases, or ergot derivative) was used was also counted as a migraine day. Details on migraine-related outcomes have been reported with the parent study (11).
Assessments
Safety was assessed by monitoring AEs and reviewing results of electrocardiograms, laboratory assessments and vital signs. AEs and vital sign data were collected throughout the OLTP and during a 12-week safety follow-up phase after the last dose. Electrocardiograms and hematology were performed at Weeks 4, 12, 24 (only for patients who increased erenumab to 140 mg at Weeks 12, 16 or 20), 36 (only for patients who increased erenumab to 140 mg at Weeks 24 or 28) and 52. Serum anti-erenumab antibodies were measured during the double-blind treatment phase (DBTP) and at Week 24 and Week 52 of the OLTP.
Efficacy was assessed using an electronic diary daily during pre-specified time intervals: For the first 12 weeks, then from Weeks 21–24, Weeks 37–40 and Weeks 49–52.
Statistical analysis
The full analysis set and safety analysis set included all patients enrolled in the study who had received at least one dose of erenumab during the OLTP. The efficacy analysis set was a subset of the full analysis set of patients who in addition had at least one change from baseline measurement for endpoint of interest.
AEs were summarized as the exposure-adjusted patient incidence rate of treatment-emergent AEs by the dose level at which the AE occurred. An AE that started in the parent study was considered treatment-emergent in the OLTP only if there was a worsening of the event. If the event continuing from the parent study had no changes in severity during OLTP, then it was not considered treatment-emergent in the OLTP. The exposure-adjusted patient incidence of a treatment-emergent AE at a dose level was defined as the number of patients with at least one reported occurrence of the event divided by the total time (patient-years) at risk for reporting the treatment-emergent AE for patients exposed. For patients with events, only the time until the first event contributed to the total patient-years, and results are presented per 100 patient-years. All other safety data, except that on anti-erenumab antibody levels, were analyzed based on the treatment received during the OLTP (70 mg only, 140 mg only or 70/140 mg).
For the analysis of efficacy endpoints, baseline was defined as the 4-week baseline period of the parent study (beginning the first day the patient used the eDiary during baseline through the day prior to study Day 1). Efficacy results presented in the OLTP are either by the combined dose group or by last dose received among completers. Summary descriptive statistics for efficacy endpoints were tabulated at each visit. No formal statistical testing was performed. Data were reported as observed, without imputation for missing data. Efficacy endpoints were summarized descriptively for all patients in the efficacy analysis set. Summary by dose group for the first 28 weeks of the OLTP was not appropriate since patients could switch dose from 70 mg to 140 mg at different time points between Week 4 and 28.
For the post-hoc efficacy analysis, analyses were performed at Weeks 40 and 52, based on the final dose received among completers; summary statistics were calculated by last dose received as collected by the electronic diary over Weeks 36–40 and 48–52, respectively. For patients switching from 70 mg to 140 mg between Weeks 4–28, by Week 40 patients would have been on 140 mg for at least 12 weeks and would have therefore achieved steady state. Consequently, by Week 52, patients would have been on 140 mg for at least 24 weeks.
Results
A total of 609 patients were enrolled, 451 (74.1%) completed the 64-week study (12 week DBTP + 52 week OLTP), and 158 (25.9%) patients discontinued the study (eight [1.3%] due to decision of the sponsor, 124 [20.4%] withdrew consent from the study, and 26 patients [4.3%] were lost to follow-up). The most common reasons for treatment discontinuation were patient request (10.5%), lack of efficacy (6.4%) and AEs (2.6%). During the study, 350 patients received erenumab 70 mg alone, 60 patients received erenumab 140 mg alone, and 199 patients had a dose increase from 70 to 140 mg erenumab by the Week 28 visit. All 609 patients received the study medication and were included in the full analysis and safety analysis sets. Figure 2 presents the study patient disposition.

Patient disposition.
At DBTP baseline, the mean (standard deviation [SD]) age of patients was 42.5 (11.3) years. Most patients were Caucasian (94.3%), and the majority of patients were women (83.6%); disease duration (mean [SD]) was 21.8 (12.4) years (Table 1). Prior preventive migraine medications were used by 454 patients (74.5%). Among those prior preventive medication users, the most frequently used medications were topiramate (68.5%), beta blockers (54.0%), and tricyclic antidepressants (48.9%); 92.3% discontinued preventive medication due to treatment failure, whether due to lack of efficacy or poor tolerability.
Patient demographics and disease characteristics at DBTP baseline (full analysis set).
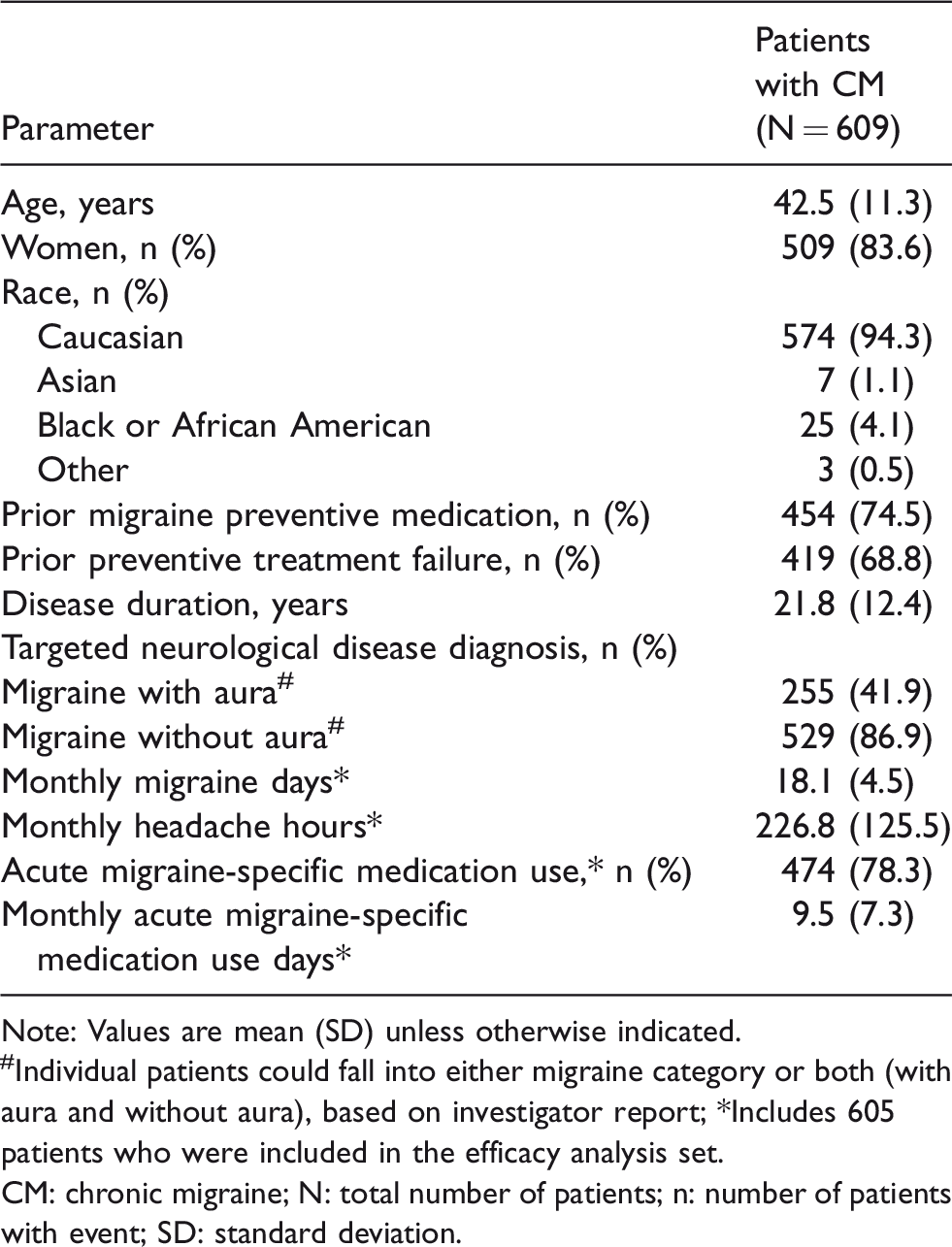
Note: Values are mean (SD) unless otherwise indicated.
#Individual patients could fall into either migraine category or both (with aura and without aura), based on investigator report; *Includes 605 patients who were included in the efficacy analysis set.
CM: chronic migraine; N: total number of patients; n: number of patients with event; SD: standard deviation.
Safety
Most patients (73.7%) received all 13 doses of erenumab. Exposure to the 70 and 140 mg doses combined was 527.0 patient-years.
Overall, 65.4% (398/609) of patients had treatment-emergent AEs. The exposure-adjusted patient incidence rate was 126.3/100 patient-years overall, and 132.0 and 148.5/100 patient-years, during erenumab 70 mg/140 mg exposure, respectively in the OLTP. In the DBTP, the exposure-adjusted patient incidence rates were 202.0/100 patient-years for placebo, and 250.3/100 and 282.3/100 patient-years for erenumab 70 mg and 140 mg, respectively; these values were slightly higher than those observed in the OLTP. The most common treatment-emergent AEs are listed in Table 2. The AE with the highest incidence among all erenumab-treated patients was viral upper respiratory tract infection (16.4/100 patient-years in the OLTP, Table 2). The frequencies of the most common AEs were comparable between erenumab and placebo during the DBTP. Most AEs were grade 1 or grade 2 in severity. Treatment-emergent AEs of grade 4 severity or fatal AEs were not observed in any patient.
Exposure-adjusted patient incidence rates (i.e. patients/100 patient-years) of treatment-emergent AEs (summarized based on the dose received when the AE occurred) (safety analysis set).

#The numbers for the two dose groups (N = 549, N = 259) are not additive to the total (N = 609), as 199 patients were exposed to both doses, and are represented in both columns depending on the dose level at which the AE occurred.
*Events with ≥4.2 patients per 100 patient-years in the total erenumab group during open-label treatment phase; time at risk during the study is the time from first dose of erenumab through to onset of first event or the minimum (end-of-study date, last dose date + 112).
Note: Grading categories determined using CTCAE version 4.03; preferred terms coded using MedDRA v20.0.
AE: adverse event; CTCAE: common terminology criteria for AEs; DBTP: double-blind treatment phase; MedDRA: Medical Dictionary for Regulatory Activities; N: total number of patients; n: number of patients with an event; OLTP: open-label treatment phase; r: exposure-adjusted patient incidence rate per 100 patient-years (n/e*100), where e, sum across all patients, the total time at risk in the study in years.
The overall exposure-adjusted patient incidence of treatment-emergent serious AEs was lower during the OLTP than was observed during the DBTP (3.8/100 patient-years [24 patients] with erenumab overall in the OLTP versus 12.1/100 patient-years [six patients], 4.1/100 patient-years [two patients], and 9.5/100 patient-years [seven patients] with the erenumab 70 mg, erenumab 140 mg, and placebo, respectively, in the DBTP) (Table 2). The serious events reported in > 1 patient included migraine (n = 4, 0.6/100 patient-years), intervertebral disc protrusion (n = 3, 0.5/100 patient-years), and depression (n = 2, 0.3/100 patient-years).
During OLTP, treatment-emergent AEs led to treatment discontinuation in 16 patients (2.5/100 patient-years, Table 2) overall in the OLTP. The exposure-adjusted patient incidence of treatment-related treatment-emergent AEs was 20.5/100 patient-years (n = 114 patients); events with incidence rate > 1 patient/100 patient-years included injection site pain, nausea, dizziness, injection site erythema, and migraine. Exposure-adjusted incidence rates were similar for both erenumab doses (21.6 and 20.9/100 patient-years for erenumab 70 mg and 140 mg, respectively).
No clinically meaningful alterations were observed in laboratory values, vital signs, electrocardiogram findings, blood pressure, or heart rate at any post-baseline time point, which is consistent with observations during the DBTP (11).
Of the 587 patients with a post-baseline result, anti-erenumab binding and neutralizing antibodies were observed in 34 (5.8%) and three (0.5%) patients, respectively. Of these patients, 17/34 and 2/3 showed transient anti-erenumab antibodies; all tested negative at the last study visit. The third patient with an anti-erenumab neutralizing antibody positive refused to participate in further neutralizing antibody follow-up tests, and so it is unknown whether they returned to antibody negativity. The neutralizing antibodies did not show any effect on efficacy.
Efficacy
In the combined dose group, the mean (SD) MMD at parent study baseline was 18.1 (4.5) days, and the mean (95% CI) change from parent study baseline at Weeks 40 and 52 was −8.7 (−9.3, −8.1) reducing MMD to 9.4 days and −9.3 (−10.0, −8.6) reducing MMD to 8.8 days, respectively (Figure 3(a)). The proportion of patients achieving a ≥50% reduction from parent study baseline in MMD at Weeks 40 and 52 was 55.6% and 59.0%, respectively (Figure 4(a)). The proportion of patients achieving a ≥75% reduction from parent study baseline in MMD was 28.1% at Week 40 and 33.2% at Week 52 (Figure 4(a)). The corresponding values for a 100% reduction from parent study baseline in MMD were 7.4% at Week 40 and 8.9% at Week 52 (Figure 4(a)). The overall mean (SD) MSMD at parent study baseline was 9.5 (7.3), and the overall mean (95% CI) change from parent study baseline was −4.6 (−5.1, −4.1), reducing the MSMD to 4.9 days at Week 40 and −5.0 (−5.5, −4.4), reducing the MSMD to 4.5 days at Week 52.

The change from baseline in number of monthly migraine days (MMD) (a) during the parent study and the OLE study; that is, DBTP and OLTP (efficacy analysis set), (b) during the OLTP at Weeks 40 and 52 by last dose received.

The proportion of patients with a ≥ 50%, ≥ 75% and 100% reduction from baseline in number of monthly migraine days (MMD) (a) during the parent study and the OLE study; that is, DBTP and OLTP (efficacy analysis set), (b) during the OLTP at Weeks 40 and 52 by last dose received.
Post-hoc analyses
At both, 40 and 52 weeks, a greater benefit was observed with the erenumab 140 mg dose compared with the 70 mg dose. The mean (SD) MMD at parent study baseline was 17.9 (4.4) and 17.8 (4.6) in the erenumab 70 and 140 mg groups, respectively. These were the long-term treatment completers who received erenumab 70 mg and 140 mg for at least 12 weeks at Week 40 and at least 24 weeks for Week 52. The mean change from parent study baseline was greater with erenumab 140 mg than with 70 mg, with an additional reduction of 2 migraine days/month at both Week 40 and Week 52 (Figure 3(b)). The reduction in MMD from parent study baseline was −7.8 days for erenumab 70 mg to 10.1 days, and −10.0 days to 7.8 days for erenumab 140 mg at Week 40. The reduction in MMD from parent study baseline was −8.5 days to 9.4 days for the 70 mg dose and −10.5 days to 7.3 days for the 140 mg dose at Week 52.
Similarly, the ≥50%, ≥75% and 100% MMD responder rates were higher with the 140 mg dose than with the 70 mg dose among completers (Figure 4(b)). At Week 40, 47.4% of the 70 mg group and 67.4% of the 140 mg group reported a ≥50% reduction in MMD; at Week 52, 53.3% of the 70 mg and 67.3% of the 140 mg group had a ≥50% reduction in MMD.
The numerical difference between the 70 mg and 140 mg responder rates was also seen for the ≥75% responder rates. At Week 40, 24.1% of the 70 mg group and 33.7% of the 140 mg reported a ≥75% reduction in MMD. At Week 52, 27.1% of the 70 mg group and 41.8% of the 140 mg group had achieved a ≥75% reduction in MMD (Figure 4(b)).
Finally, the 100% responder rate was nearly two-fold higher with the 140 mg dose at both Weeks 40 and 52 than with the 70 mg dose. At Week 40, 4.8% of the 70 mg group and 10.7% of the 140 mg group reported at least a month of no migraines (i.e. a 100% decrease in MMD). At Week 52, 6.1% of the 70 mg group and 12.7% of the 140 mg group had at least a month of no migraines (Figure 4(b)).
The mean (SD) monthly MSMD in the subset of patients using acute migraine-specific medications at parent study baseline was 12.2 (5.9) with change from parent study baseline (mean [95% CI]) reported at Weeks 40 and 52 as −5.8 (−6.3, −5.2) to 6.4 days and −6.4 (−7.0, −5.8) to 5.8 days, respectively (Figure 5(a)). Among the completers, the mean (SD) monthly acute MSMD among the subset of patients using acute migraine-specific medications at parent study baseline was 12.4 (5.5) days with the 70 mg dose and 11.3 (6.5) days with 140 mg dose. The reduction in mean MSMD was also greater with the 140 mg dose versus 70 mg dose (Figure 5(b)). At Week 40, the decrease in monthly MSMD for the 70 mg group was −5.6, reducing the monthly MSMD to 7.1 days; for the 140 mg dose group it was −6.2, reducing the monthly MSMD to 5.2 days. At Week 52, the decrease in monthly MSMD for the 70 mg group was −6.2, reducing the MSMD to 6.5 days; for the 140 mg group the decrease was −6.7, reducing the monthly MSMD to 5.0 days.
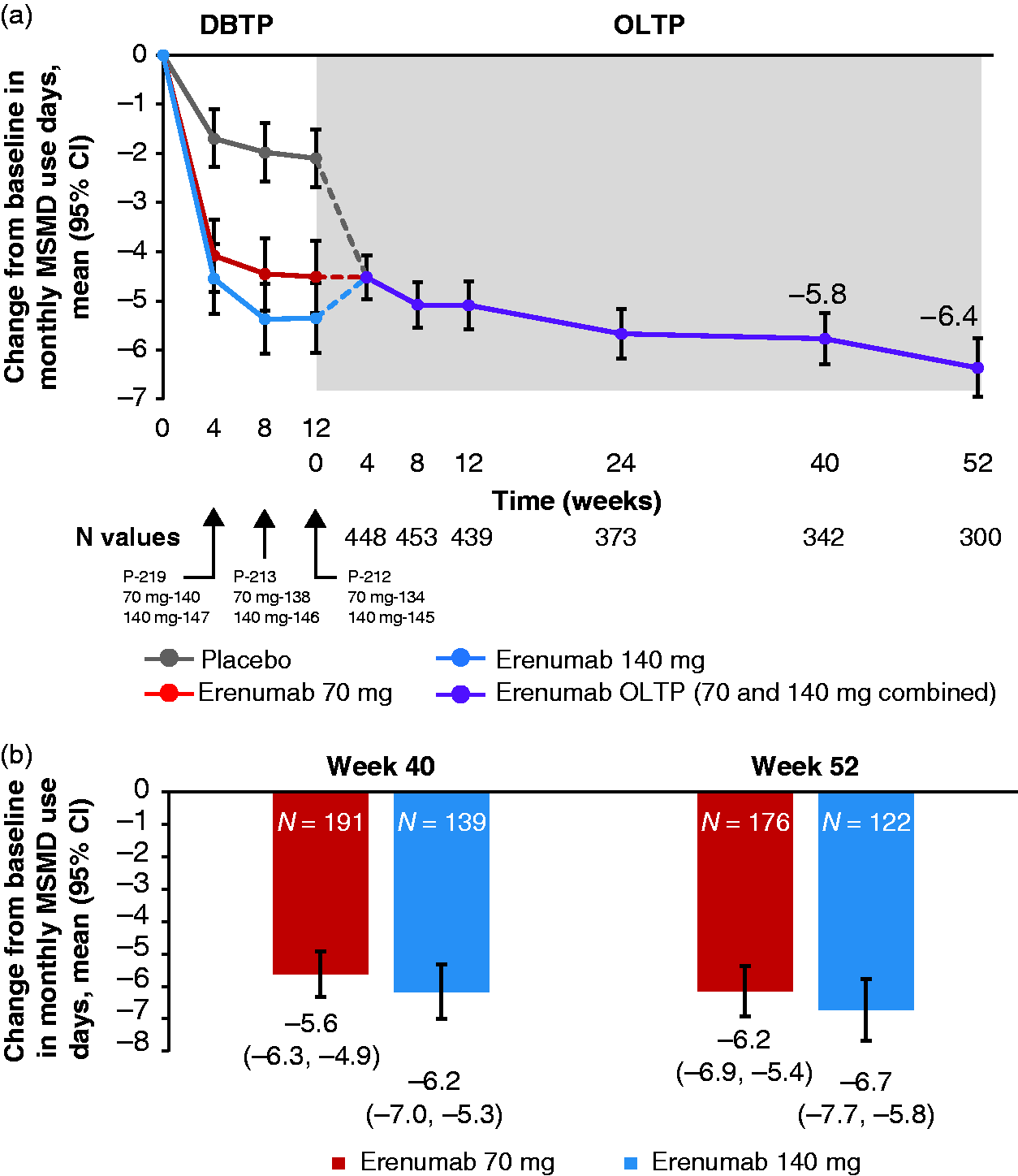
The change from parent study baseline in monthly migraine-specific medication use days (MSMD) in acute migraine-specific medication users at baseline (a) during the parent study and OLE study; that is, DBTP and OLTP (efficacy analysis set), (b) during the OLTP at Weeks 40 and 52 by last dose received.
Discussion
Results from this OLTP study demonstrate the long-term safety and efficacy of erenumab over a 52-week period in patients with CM following the 12-week DBTP. Clinical characteristics specific to migraine at baseline were consistent with a patient population with CM (11). The Summary of Product characteristics for erenumab lists constipation, itching, muscle spasms, and injection site reactions as common AEs affecting up to 1 in 10 patients. Most AEs reported in the current study were grade 1 or grade 2 in severity. Treatment-emergent AEs of grade 4 severity and fatal AEs were not reported in any patient. The safety profile of erenumab in the OLTP was consistent with that in the DBTP, with no new safety concerns emerging.
Across the placebo-controlled clinical trials of erenumab (including the DBTP of this OLTP study), there was a low incidence of binding and neutralizing anti-erenumab antibodies (10, 11). The development of anti-erenumab antibodies has not been associated with any clinical outcomes or safety events. The generation of binding and neutralizing antibodies post-baseline was transient in the majority of patients.
Erenumab treatment demonstrated sustained efficacy during the 52-week OLTP. Overall, there was a nearly 50% reduction in mean MMD and MSMD at Week 52 with erenumab treatment. This reduction in MMD means that, on average, most patients would no longer be classified as having CM. Similarly, at the individual level, more than 50% of patients achieved at least a 50% reduction in MMD at Week 52. Due to the protocol amendment allowing a dose increase of erenumab by Week 28, patients whose dose increased to 140 mg would have been on 140 mg for at least 12 weeks at Week 40, which is sufficient to have reached steady state concentrations. Thus, Week 40 provided the first opportunity to assess efficacy in the study population based on the final dose received, 70 mg or 140 mg. A post-hoc analysis of efficacy at Week 40 and Week 52 based on the last dose received showed a numerically greater benefit for the 140 mg dose over the 70 mg dose on a variety of endpoints including MMD, responder rates, and reduction in the MSMD for acute medications.
The proportion of patients with a ≥50% response was greater in the OLTP at Week 52 (67.3% and 53.3%, with 140 mg and 70 mg), compared with the DBTP (41% and 40%, respectively) (11), suggesting sustained or increased efficacy with long-term treatment, although it is possible that withdrawal of patients not responding to treatment might have impacted these results. Similarly, the reduction in MSMD at Week 52 was numerically higher with both doses (−6.2 days for the 70-mg dose and −6.7 days for the 140-mg dose) compared with the DBTP (−3.5 and −4.1 days, respectively), showing a reduction of nearly 3 monthly MSMD from Week 12 of the DBTP (11). A reduction in the MSMD is important, as this is an important risk factor for chronification of migraine (15).
While this might suggest that efficacy increases over time across endpoints, given the open-label nature of the study and the loss of patients over time, not unusual for open-label extensions, our overall interpretation is that efficacy is at least sustained throughout the study, given the possibility of bias introduced by the loss of patients who did not experience the same level of efficacy as those who remained in the study. Approximately 25.9% of patients discontinued the OLTP, and 22.8% of patients discontinued the investigational product. The number of patients who discontinued treatment as a result of AEs was small. The data from this OLTP study show sustained efficacy compared with the findings from the 12-week parent study and support a durable effect of treatment, with a safety profile at 52 weeks similar to that in the parent study at 12 weeks.
Limitations of the study include that it was non-randomized, and erenumab was administered open-label, without blinding of dose. Nonetheless, the results for efficacy endpoints favored the 140-mg dose of erenumab.
Conclusion
The results from this 52-week open-label extension study demonstrate the long-term safety and tolerability of erenumab in patients with CM. The spectrum of AEs was similar to that observed during the placebo-controlled study, with no new safety concerns and no increase in the rate of AEs. Efficacy was sustained throughout the 52-week period.
A numerically greater efficacy was observed with the erenumab 140 mg versus 70 mg dose on a variety of endpoints including reduction in mean MMD, responder rates, and reduction in MSMD for use of acute medications, suggesting erenumab 140 mg may provide better long-term efficacy in CM than 70 mg. Overall, the favorable safety, tolerability, and efficacy profile in this study, and its once-monthly administration, may mean that subcutaneous erenumab has the potential to increase real-world adherence to treatment in patients with chronic migraine.
Clinical implications
This 52-week open-label extension study demonstrates the long-term safety and tolerability of erenumab in patients with chronic migraine. Adverse events reported during the open-label treatment phase were similar to those observed during the double-blind treatment phase, with no new safety events and no increase in adverse events. Erenumab 140 mg showed greater clinical benefit than the 70 mg dose in this study, as demonstrated by post-hoc analyses of efficacy parameters, including reductions in mean monthly migraine days, responder rates, and reduction in days of use of acute migraine-specific medications. Although dropouts occurred, as is typical in open-label extension trials, this study demonstrated at least sustained benefit for trial completers using erenumab across 52 weeks. The favorable safety, tolerability, and efficacy profile for erenumab demonstrated in this study supports the prolonged use of erenumab for migraine prophylaxis.
Supplemental Material
CEP912726 Supplemental material - Supplemental material for Long-term safety and efficacy of erenumab in patients with chronic migraine: Results from a 52-week, open-label extension study
Supplemental material, CEP912726 Supplemental material for Long-term safety and efficacy of erenumab in patients with chronic migraine: Results from a 52-week, open-label extension study by Stewart J Tepper, Messoud Ashina, Uwe Reuter, Jan Lewis Brandes, David Doležil, Stephen D Silberstein, Paul Winner, Feng Zhang, Sunfa Cheng and Daniel D Mikol in Cephalalgia
Footnotes
Previous presentations
Data from this study was presented at the 60th Annual Scientific Meeting of the American Headache Society (AHS), 28 June to 1 July, 2018, San Francisco, California, USA.
Contributors
All authors participated in the study design, implementation and/or conduct of the study. All authors contributed to the review of the protocol and approved the final manuscript.
Acknowledgements
Declaration of conflicting interests
The authors declare the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ST was an employee of Cleveland Clinic during this study and has research grants (no personal compensation) from Alder, Allergan, Amgen, ATI, Dr. Reddy’s, ElectroCore, eNeura, Neurolief, ScionNeurostim, Teva, and Zosano; he has consulted for Acorda, Alder, Alexsa, Allergan, Alphasights, Amgen, ATI, Axsome Therapeutics, Biohaven, Charleston Labs, DeepBench, Dr. Reddy’s, ElectroCore, Eli Lilly, eNeura, ExpertConnect, Gerson Lehman Group, Guidepoint Global, GSK, Impel, Magellan Rx Management, Navigant Consulting, Neurolief, Nordic BioTech, Novartis, Pfizer, Reckner Healthcare, Satsuma, ScionNeurostim, Slingshot Insights, Sorrento, Sudler and Hennessey, Supernus, Teva, Theranica, Trinity Partners, XOC, and Zosano; he has stock options for ATI; receives a salary from the American Headache Society; and royalties from Springer.
MA is consultant or scientific advisor for Allergan, Amgen, Alder, Eli Lilly, Lundbeck, Novartis, and Teva; primary investigator for Allergan, Amgen, Eli Lilly, ElectroCore, Novartis and Teva; has received grants from Lundbeck Foundation, Novo Nordisk Foundation, and a research grant from Novartis.
UR has received consulting fees, speaking/teaching fees, and/or research grants from Allergan, Amgen, Autonomic Technologies, CoLucid, ElectroCore, and Novartis.
JLB received consulting fees, speaking/teaching fees, and/or research grants from Allergan, Amgen, Teva, Eli Lilly, Alder, Lundbeck, Supernus, Clinvest, Theranica and Biohaven.
DD received consulting fees and/or advisory panel member and speaking and/or teaching fees from Allergan, Amgen, Biogen Idec, Novartis, Bayer, and Teva.
SS was a consultant and/or advisory panel member, and received honoraria from Alder Biopharmaceuticals, Allergan, Amgen, Avanir, Dr. Reddy’s, eNeura, ElectroCore Medical, LLC, Medscape LLC, Medtronic, Mitsubishi Tanabe Pharma America, Inc., NINDS, Supernus Pharmaceuticals, Inc., Trigemina, and Teva.
PW received consulting fees/honoraria from Allergan, Amgen, Lilly, Teva and Supernus; speaker’s bureau: Allergan, Amgen, Avanir, Teva and Supernus; research grants: Allergan, Amgen, NuPathe, AstraZeneca, Avanir, Eli Lilly, Novartis and Teva
SC, FZ, and DDM, employees and stocks: Amgen.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by Amgen, Thousand Oaks, CA, USA, and Novartis Pharma AG, Basel, Switzerland. Employees of the sponsors were involved in study design, data collection, analysis and interpretation. All authors, including those who are employees of the study sponsors, drafted and/or revised the manuscript and approved the final version for submission.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.