
Abstract
Background
Ubrogepant is a novel, oral calcitonin gene–related peptide (CGRP) receptor antagonist in development for the acute treatment of migraine. This trial evaluated the safety and tolerability of ubrogepant, focusing on hepatic safety, when administered intermittently with high-frequency dosing to healthy participants.
Methods
In this phase 1, multicenter, double-blind, parallel-group trial, healthy adults (age 18–50 years) were randomized 1:1 to placebo or ubrogepant. Ubrogepant was dosed at 100 mg (2 × 50 mg tablets) on 2 consecutive days followed by 2 consecutive days of placebo, alternating for 8 weeks. Primary outcome measures were safety and tolerability.
Results
Of participants randomized (n = 518), 516 were included in the safety population (n = 260 placebo; n = 256 ubrogepant). Treatment-emergent adverse events were reported in 45% of placebo and 44% of ubrogepant participants. The most common was headache (10% placebo; 11% ubrogepant). Overall, seven cases of alanine aminotransferase and/or aspartate aminotransferase levels ≥ 3 × the upper limit of normal (five placebo, two ubrogepant) were reported and adjudicated by a panel of independent liver experts blinded to treatment. Four cases were judged unlikely related to treatment. Two cases (one placebo, one ubrogepant) were judged possibly related, and one (ubrogepant) probably related. Alanine aminotransferase increases to ≥ 3 × the upper limit of normal in the two ubrogepant cases (possibly or probably related) were transient and resolved with continued dosing; both cases were asymptomatic, with no concurrent bilirubin elevation.
Conclusion
Ubrogepant was well tolerated following intermittent, high-frequency dosing in healthy participants, with no clinically relevant signal of hepatotoxicity.
Trial Registration
NA.
Introduction
Calcitonin gene–related peptide (CGRP) is a neuropeptide that plays an integral role in migraine attacks (1,2). CGRP receptor antagonism has proven to be effective in acute treatment of migraine attacks (3–7). Development of two of the first-generation small-molecule CGRP receptor antagonists, telcagepant (MK-0974) and MK-3207, was discontinued because clinical data suggested a potential for drug-induced liver injury (4,8). Known mechanisms of drug-induced liver injury include oxidative stress, development of reactive metabolites, mitochondrial toxicity, altered bile acid homeostasis, and innate and adaptive immune dysfunction (9). Liver test abnormalities associated with telcagepant and MK-3207 were attributed to molecule-specific metabolites, rather than a class effect due to CGRP inhibition (8,10). The lack of any observed liver enzyme abnormalities with CGRP pathway blockade by monoclonal antibodies further supported this conclusion (11,12).
Ubrogepant is a small-molecule, orally delivered, potent, and specific CGRP receptor antagonist that is under investigation for the acute treatment for migraine. It was designed to be more potent than previous CGRP receptor antagonists, with the goal of decreasing the dose needed for therapeutic effect and the associated risk of hepatotoxicity. In functional assays, ubrogepant potently blocked human α-CGRP–stimulated cAMP responses (IC50 of 0.08 nM) in human CGRP receptor–expressing HEK293 cells and exhibited highly selective antagonist activity for the CGRP receptor compared with other members of the human calcitonin receptor family (13,14). This phase 1 trial evaluated the safety and tolerability of ubrogepant, focusing on hepatic safety, when administered intermittently with high frequency to healthy participants. The data have been presented in preliminary form (15).
Methods
Trial design and participants
This phase 1, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial in healthy adults consisted of a 4-week screening period followed by an 8-week (56-day) double-blind treatment period and a 4-week safety follow-up period (Figure 1). The trial was conducted at six trial centers in the United States between 12 November 2017 and 17 May 2018. The trial protocol was approved by appropriate institutional review boards (IRBs) for each trial center (IntegReview IRB, Austin TX, USA or Advarra [previously Chesapeake] IRB, Columbia, MD, USA). Informed written consent was obtained from all participants.
Trial design.
Eligible participants were healthy males or females aged 18 through 50 years (inclusive) with a body mass index between 18 and 30 kg/m2. Participants were required to have normal ( < 1 × the upper limit of normal [ULN]) levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin at each of two screening visits separated by at least 14 days. Participants were excluded if they had any of the following: any clinically significant disease state (including migraine); a history of hypersensitivity or clinically significant adverse reaction to a CGRP receptor antagonist; sitting systolic blood pressure ≥ 140 or ≤ 90 mm Hg or diastolic blood pressure ≥ 90 or ≤ 50 mm Hg at initial screening or before receiving trial medication on day 1; potentially clinically significant abnormal electrocardiogram results; QT interval prolongation; sitting pulse rate < 40 bpm or > 100 bpm; any medication use (including over-the-counter products, hormonal contraceptives, supplements, herbal preparations, or products containing grapefruit) during the screening period; or had participated in a trial of ubrogepant or another CGRP receptor antagonist within 3 months. For current smokers, daily smoking was prohibited. Occasional smoking was permitted and limited to no more than 8 cigarettes on any smoking day. Occasional smoking was allowed because ubrogepant is not metabolized by CYP1A2 or CYP1A1 (which could be induced in the presence of smoking). Participants must have abstained from drinking alcohol from the time the informed consent was signed through the end of the study.
Treatment and procedures
Eligible participants were stratified by sex and randomly assigned (1:1; block size = 4) to receive placebo (two placebo tablets) or ubrogepant 100 mg (2 × 50 mg tablets) using an automated interactive Web response system in a double-blind fashion based on a computer-generated randomization scheme. Starting on day 1, participants in the ubrogepant group received 2 consecutive days of ubrogepant 100 mg followed by 2 days of placebo, alternating for a total of 56 days (8 weeks). Participants in the placebo group received two tablets of matching placebo daily for 56 days (8 weeks). Participants and staff were blinded to treatment assignment. The two trial treatments were indistinguishable and administered in the same manner.
Randomized participants returned to the trial center once or twice per week for safety assessments, on-site dosing, and blood sampling for pharmacokinetic assessments. Treatment was administered at the trial center approximately twice a week, after which blood samples for pharmacokinetic analysis were obtained at either 0.5 hours (days 1, 14, 25, 37, and 49), 1 hour (days 5, 17, 42, and 53), or 2 hours post dose (days 9, 21, 33, and 45). End-of-trial assessments were completed on or within 4 days of the last day of the double-blind treatment period (day 56) or early termination. A safety follow-up visit occurred 4 weeks after the last dose.
Safety assessments
The primary outcome measures were safety and tolerability. All adverse events (AEs) were monitored and recorded from the treatment start date to the safety follow-up visit or within 30 days after last dose for participants without the safety follow-up visit. An event that occurred after the safety follow-up visit, or more than 30 days after the last dose of study treatment for participants without the safety follow-up visit, was not considered to be treatment-emergent. Treatment-emergent AEs (TEAEs) were defined as AEs occurring after the first dose of treatment or AEs that existed before the first dose but increased in severity after the first dose. Adverse events were coded using the Medical Dictionary for Regulatory Activities version 20.1. Additional safety assessments included clinical laboratory tests, vital sign measurements, electrocardiogram, physical examination, and the Columbia–Suicide Severity Rating Scale (16). To closely monitor hepatic safety, serum chemistry values were obtained at both screening visits and on days 1, 5, 14, 21, 28, 33, 42, 49, 56 (end of trial), and 84. All clinical laboratory tests were determined by a central laboratory (ACM Global Laboratories, Rochester, NY).
Selected TEAEs designated as events of clinical interest were monitored. The events of clinical interest included treatment-emergent suicidal ideation (i.e. type 4 or 5 on the Columbia–Suicide Severity Rating Scale) or any suicidal behavior; treatment-emergent elevation of ALT and/or AST value ≥ 3 × ULN; and all potential Hy's Law cases, defined as an ALT or AST value ≥ 3 × ULN accompanied by a total bilirubin value ≥ 2 × ULN and an alkaline phosphatase (ALP) value < 2 × ULN, all based on blood draws collected within a 24-hour period. A blinded, independent clinical adjudication committee of three liver experts reviewed each post-treatment case of ALT or AST ≥ 3 × ULN to assess the relationship to study treatment and the presence of any potentially confounding factors. The ULN for ALT was 44 U/L in males and 33 U/L in females. The ULN for AST was 39 U/L in males and 34 U/L in females. The ULN for ALP was 129 U/L in males and 98 U/L in females. The ULN for total bilirubin was 1.2 mg/dL in males and females. Causality was graded using a 3-point scale (probable: ≥50% likelihood; possible: 25–49% likelihood; unlikely: <25% likelihood).
Statistical analyses
The sample size was driven by regulatory recommendations for the duration and number of participants exposed rather than statistical considerations. By randomizing 500 participants in a 1:1 ratio to placebo or ubrogepant 100 mg (250 participants in each group), it was anticipated that at least 200 participants in the ubrogepant 100 mg group would complete the 8-week double-blind treatment period.
Of all participants randomized to a treatment group, those who received at least one dose of study treatment were included in the safety population analysis. The incidence of TEAEs and descriptive statistics for the various safety measures were summarized by treatment group.
Results
Participant characteristics
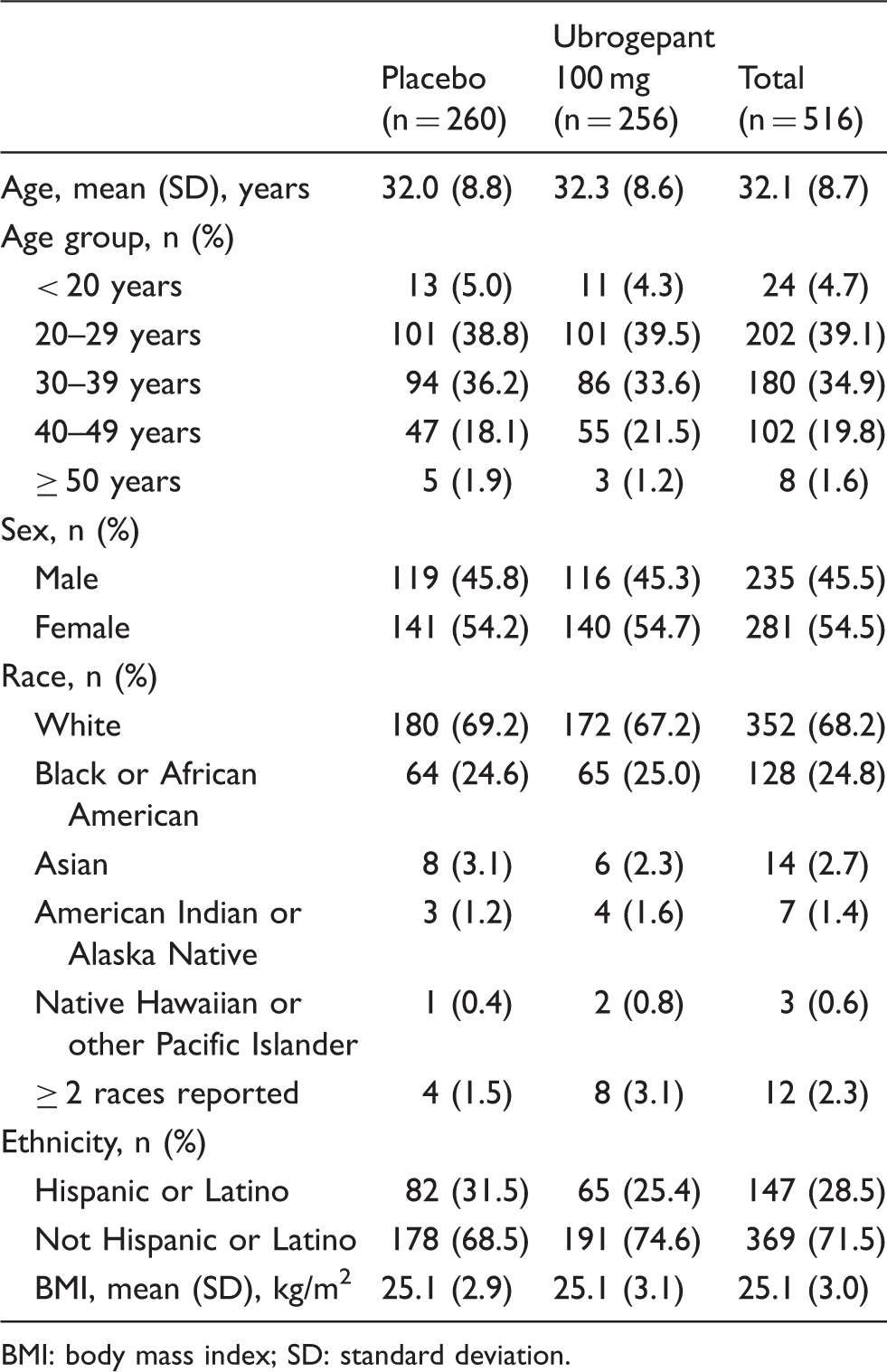
Of the 518 participants who were randomized, 516 participants (n = 260 placebo; n = 256 ubrogepant) received at least one dose of study treatment during the double-blind period and were included in the safety analysis. Baseline characteristics were similar between the placebo and ubrogepant groups (Table 1). Participants were primarily female (54%) and white (68%); and the mean age of participants in both groups was 32 years. Mean body mass index (BMI) was 25.1 kg/m2. Overall, 468 of 516 participants (91%) completed the 8-week double-blind treatment period (234 participants in each treatment arm). A total of 26 (10%) participants in the placebo group, and 22 (9%) in the ubrogepant group discontinued the trial early (Figure 2).
Participant disposition. Demographics and baseline characteristics. BMI: body mass index; SD: standard deviation.
Pharmacokinetics
Ubrogepant 100 mg resulted in adequate systemic exposure, with mean (SD) plasma concentrations of 148 ng/mL (134 ng/mL), 318 ng/mL (214 ng/mL), and 292 ng/mL (149 ng/mL) at 0.5 hours, 1 hour, and 2 hours post dose, respectively.
Safety
Overall summary of adverse events.

AE: adverse event; TEAE: treatment-emergent adverse event; SAE: serious adverse event.
TEAEs were defined as events that initially occurred or increased in severity on or after the first dose of treatment. Events that occurred after the safety follow-up visit for participants with safety follow-up visit data or > 30 days after the last dose of treatment for participants without safety follow-up visit data were not considered to be treatment-emergent.
SAEs were defined as events that occurred between the treatment start date and the safety follow-up visit, or within 30 days after the last dose of treatment for participants without safety follow-up visit data.
Discontinuation events that occurred between the treatment start date and the safety follow-up visit, or within 30 days after the last dose of treatment for participants without safety follow-up visit data.
Hepatic safety
Hepatic laboratory parameters.

ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; SD, standard deviation; ULN: upper limit of normal.
Participants with AST or ALT values ≥ 3 × ULN
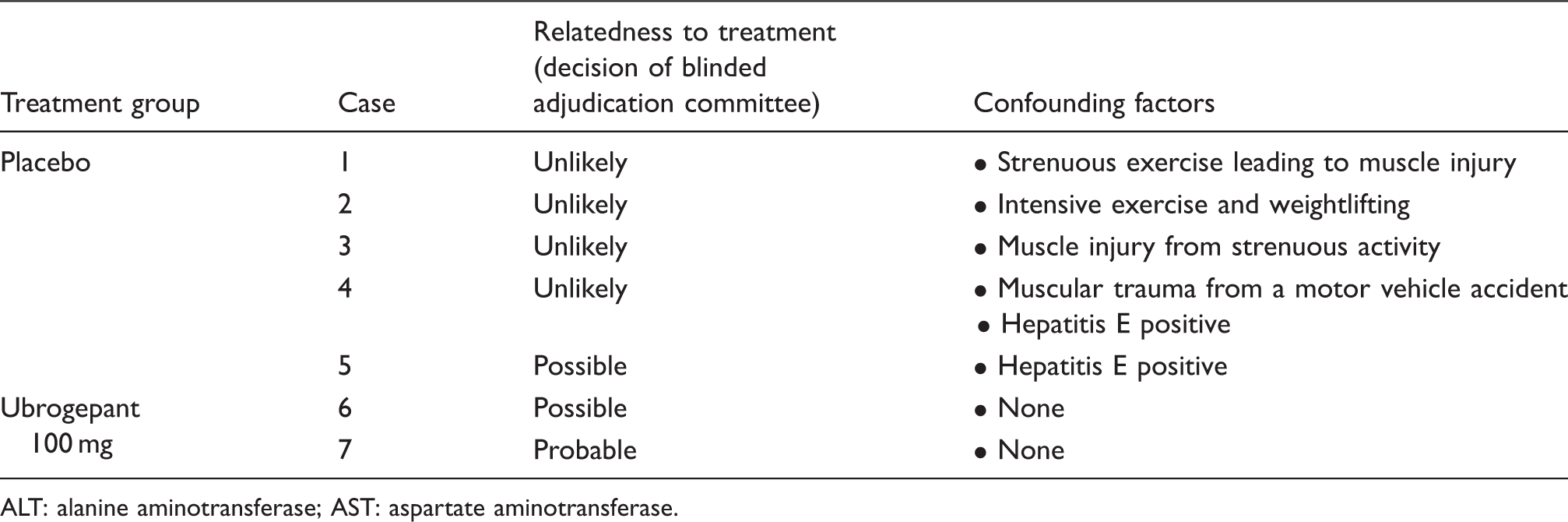
A total of seven participants (placebo n = 5; ubrogepant n = 2) had postbaseline ALT and/or AST values ≥ 3 × ULN. Each of these cases was reviewed by the blinded clinical adjudication committee to assess causality and potential confounding factors (Table 4). Four of the five cases in the placebo group were judged unlikely to be related to treatment, and one case was deemed possibly related. Among the ubrogepant recipients, one of the cases of ALT values ≥ 3 × ULN was judged possibly related (Figure 3(a)), and one was judged by majority vote to be probably related to treatment (Figure 3(b)). Both cases in the ubrogepant group were asymptomatic, with no concurrent bilirubin elevations, and resolved with continued ubrogepant dosing. Extensive aggregate review of all cases with the adjudicators after unblinding of the data at the end of the trial concluded that no hepatic safety concerns were identified for ubrogepant.
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values over time in the two ubrogepant-treated participants with ALT or AST values ≥ 3 × the upper limit of normal (ULN) during double-blind treatment. The first case (a) was in a 29-year-old female participant randomized to ubrogepant 100 mg who reported no relevant medical history or concomitant medications and had normal levels of ALT, AST, blood creatine phosphokinase (CPK), alkaline phosphatase (ALP), and total bilirubin at baseline. The second case (b) was in a 26-year-old female participant randomized to ubrogepant 100 mg who reported no relevant medical history or concomitant medications and had normal ALT, AST, CPK, ALP, and total bilirubin levels at baseline. Note that the second rise in serum aminotransferases occurred after discontinuation of ubrogepant, and there was a negative rechallenge. For this reason, one of the three hepatologists thought that the event was only possibly related to ubrogepant treatment. INR: international normalized ratio. Findings of the blinded adjudication committee of liver experts regarding the causality of ALT or AST values ≥ 3 × the upper limit of normal. ALT: alanine aminotransferase; AST: aspartate aminotransferase.
Discussion
This placebo-controlled study closely monitored the safety and tolerability of ubrogepant 100 mg when dosed at the frequency of 2 days on followed by 2 days off for 8 weeks in healthy participants, showing no significant potential for hepatic injury. The intermittent dosing frequency of 2 days of ubrogepant followed by 2 days of placebo was selected to reflect the episodic nature of migraine and achieve high-frequency dosing and drug exposure (i.e. 28 days of drug exposure during a 56-day treatment period). The treatment duration of 8 weeks with a 4-week safety follow-up period in the current trial provided sufficient time to monitor for delayed hepatic AEs; this design was based on results from the telcagepant clinical trials, during which all cases of AST and ALT elevations occurred within 8 weeks of treatment initiation (8). Hepatic laboratory values were monitored approximately weekly throughout the 8-week double-blind treatment period to provide a high likelihood of detecting hepatic signals if the studied dosing were associated with any hepatic toxicity.
Results demonstrated that ubrogepant had minimal effects on hepatic laboratory values, with infrequent elevations in aminotransferases to ≥ 3 × ULN. The two cases of transient aminotransferase elevations to ≥ 3 × ULN in the ubrogepant group were asymptomatic, with no concurrent bilirubin elevations. Importantly, the aminotransferase elevations resolved with continued ubrogepant dosing. These findings are consistent with those of a long-term safety trial, in which adults who treated up to eight migraine attacks every 4 weeks showed no hepatic safety concerns (17).
It is important to note that transient postbaseline elevations in ALT and/or AST ≥ 1 × ULN occurred in 27% of participants in the placebo and ubrogepant groups of the current trial, suggesting that such fluctuations are common in a population of healthy individuals. Given the overall balance in aminotransferase elevations between placebo and active treatment, these observed elevations were likely to have been fluctuations unrelated to the study drug. Overall, no safety concerns were identified with the high-frequency dosing of ubrogepant.
One limitation of this trial is that participants were healthy individuals with BMI ≤ 30 kg/m2, and they were not allowed to take any other medications during the trial. Therefore, the findings may not represent outcomes in obese patients who may have underlying liver abnormalities (e.g. fatty liver) or in patients with migraine who may be on multiple concomitant medications, including those with potential for liver injury (e.g. acetaminophen/paracetamol). In addition, because of the limited timing of pharmacokinetic sample collection, full pharmacokinetic parameters were not calculated. However, as summarized above, the overall pharmacokinetic, safety, and hepatic safety results observed in this trial are consistent with results of a 52-week, long-term safety trial in a migraine population (17–19).
Conclusion
Findings from this phase 1, double-blind, placebo-controlled safety trial in healthy participants demonstrate that intermittent dosing of ubrogepant (2 days of ubrogepant 100 mg alternating with 2 days of placebo) was not associated with an increase in ALT or AST compared with placebo, or with the development of any clinically significant liver injury. When administered at the studied frequency, ubrogepant was safe and well tolerated over an 8-week period, with no clinically relevant signal of hepatotoxicity.
Footnotes
Clinical implications
Ubrogepant was well tolerated in healthy participants, with no identified safety concerns.
Intermittent dosing of 2 days of ubrogepant 100 mg alternating with 2 days of placebo was not associated with persistent increases in AST or ALT compared with placebo.
Overall, ubrogepant showed no clinically relevant signal of hepatotoxicity with intermittent dosing.
Acknowledgements
Thank you to all the participants and investigators who participated in this study. Participants received from $3250 up to $6000 for their participation in the trial; the amount varied across each of the six trial centers. Medical writing and editorial support was provided by Lela Creutz, PhD, and Lisa Feder, PhD, of Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and funded by Allergan plc. The opinions expressed in this article are those of the authors. The authors received no honorarium/fee or other form of financial support related to the development of this article.
List of investigators and trial centers
Dr. Jeffrey Peterson, Altasciences/Algorithme Pharma USA, 4837 Amber Valley Parkway, Fargo, ND 58104; Dr. Martin Kankam, Vince & Associates Clinical Research, Inc., 10103 Metcalf Avenue, Overland Park, KS 66212; Dr. Terry O’Reilly, Celerion, 2420 W. Baseline Rd., Tempe, AZ 85283; Dr. Philip Mathew, Celerion, 621 Rose Street, Lincoln, NE 68502; Dr. Samer Nakhle, PPD, Las Vegas Clinical Research Unit, 8285 W. Arby Ave, Suite 331, Las Vegas, NV 89113; Dr. Rebecca Wood-Horrall, PPD Phase I Clinic, 7551 Metro Center Drive, Suite 200, Austin, TX 78744.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PJG has received personal fees from Alder Biopharmaceuticals, Allergan plc, Autonomic Technologies Inc., Electrocore LLC, eNeura, Impel Neuropharma, Massachusetts Medical Society, MedicoLegal work, MundiPharma, Novartis, Teva Pharmaceuticals, Trigemina Inc., and Up-to-Date; has received grants and personal fees from Amgen and Eli-Lilly; and holds a patent for magnetic stimulation for headache assigned to eNeura without fee. SJT has served as a consultant for Acorda, Alder, Alexsa, Allergan plc, Alphasights, Amgen, ATI, Axsome Therapeutics, BioDelivery Sciences International, Biohaven, Charleston Labs, Decision Resources, DeepBench, Dr. Reddy’s, ElectroCore, Eli Lilly, eNeura, GLG, GSK, Guidepoint Global, Impel, M3 Global Research, Magellan Rx Management, Medicxi, Navigant Consulting, Neurolief, Nordic BioTech, Novartis, Pfizer, Reckner Healthcare, Relevale, Satsuma, Scion Neurostim, Slingshot Insights, Sorrento, Sudler and Hennessey, Teva, Theranica, Thought Leader Select, Trinity Partners, XOC, and Zosano. His employer, Dartmouth-Hitchcock Medical Center, receives research grants from Alder, Allergan plc, Amgen, ATI, Dr. Reddy’s, Scion Neurostim, Neurolief, Teva, and Zosano, receives a salary as Editor-in-Chief of Headache Currents from the American Headache Society (AHS), and receives royalties for books published by Springer. PW has served as a compensated consultant to Allergan but received no compensation for his involvement as an author of this publication. He has also served as a consultant for Abivax, AbbVie, Accorda, Actelion, Agios, Akebia, Allergan, Almirall, Alnylam, Amgen, Astellas, Axovant, Belhus, Biocryst, Biogen, Biohaven, BMS, Cytier, Debiopharm, Diaichi-Sankyo, DILIsym Services, Inc., ERX, Ferring, Frazier, Genentech, Gilead, GW, Hoffman LaRoche, HRA, Indalo, Intercept, F2G, Janssen, KBP Biosciences, Novartis, Palladia, Pfizer, PTC, Receptos, Seattle Genetics, Sojournix, Spark Therapeutics, Strongbridge, Sumitomo Dianippon, and the TB Alliance. GA, RM, MB, LS, MF, AS, JMT, and AJ are employees of Allergan plc and may own stock/options in the company.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This trial was sponsored by Allergan plc, Dublin, Ireland.