
Abstract
Background and objectives
Ictal epileptic headache is a rare form of painful seizure, habitually consisting of migrainous or tension-type headache. We describe a case of a patient with short-lasting, severe retroorbital pain attacks caused by frontal lobe epilepsy.
Case report
A 25-year-old male patient presented with recurrent attacks of paroxysmal, short-lasting, excruciating left periorbital and facial pain mainly occurring from sleep. After intracranial EEG exploration and resection of a right prefrontal focal cortical dysplasia, long-term seizure and headache remission was obtained.
Discussion
Our case extends the clinical and neuroanatomical spectrum of ictal epileptic headache and suggests that long-term remission can be obtained by resective epilepsy surgery. It also reinforces the role of the prefrontal cortex in the pain matrix and pain generation.
Conclusion
Despite its rarity, ictal epileptic headache should be suspected in selected patients, particularly those with other ictal symptoms and signs, history of epileptic seizures, or neuroimaging abnormalities.
Introduction
Despite the high comorbid rate of chronic epilepsy and headache, ictal epileptic headache (IEH) remains an extremely rare and seldom-reported condition. The third edition of the International Classification of Headache Disorders defines IEH as headache occurring simultaneously with onset of a partial seizure, and either ipsilateral to the ictal discharge or spontaneously improving or remitting immediately after seizure termination (1). Although it usually presents with migrainous or tension-type features, some cases mimic less typical headache syndromes, such as trigeminal autonomic cephalalgia (TAC) (2) and trigeminal neuralgia (3). We report a case of IEH initially diagnosed as trigeminal neuralgia, successfully treated with resective brain surgery.
Case report
A 25-year-old right-handed male patient presented with a history of paroxysmal headache attacks since age nine.
He described these episodes as abrupt onset of severe, excruciating, stabbing pain behind his left eye, “as if someone is stabbing me”, radiating seconds later to his left hemiface. Some attacks would be so severe that he would describe them as “the most severe pain” he had ever experienced. The most intense attacks would occasionally be followed by short-lasting twitches of his left face and arm, although the patient was not sure whether these were involuntary or his reaction to the extreme pain. In the aftermath of many episodes he would feel a residual burning sensation in his left face, sometimes lasting for hours, and would become drowsy.
Attacks most frequently occurred spontaneously and repeatedly during the night, waking the patient up and significantly disrupting his sleep and causing daytime somnolence. With time, the attacks became shorter but more severe, lasting about 10 seconds, and occurring up to 20 times per night. Studying aeronautical engineering at the time, he temporarily suspended his studies due to the disabling nature of the attacks.
The patient denied excessive tearing, conjunctival hyperaemia, sudoresis, miosis, ptosis, or rhinorrhoea. He did not recognize any particular triggers, including touching the face. He also denied any sensation of fear, palpitations, hypersalivation, speech difficulty or loss of awareness during the attacks.
There was no history of febrile convulsions, head trauma or central nervous system infection. There was no family history of epilepsy, chronic headache or other neurological conditions. He did not smoke or drink alcohol.
His initial diagnosis as a child was of trigeminal neuralgia; however, at 14, during an EEG study in another centre, a convulsive seizure was recorded, reporting right hemispheric ictal discharges, and a diagnosis of epilepsy was made.
He had been previously treated with multiple anticonvulsants, including levetiracetam, carbamazepine, gabapentin, clobazam and phenytoin, without significant improvement. At the time of our evaluation he was being treated with lacosamide (350 mg/day) and eslicarbazepine (2 g/day).
A scalp video-EEG telemetry documented 13 attacks in three nights. The patient would quickly arouse from sleep, complain of severe pain and at times swear and become restless. The attacks lasted up to 20 seconds, and despite antiepileptic drug reduction, no clear ictal EEG changes were recorded.
1.5 Tesla Brain MRI (Figure 1(a)) showed two right-hemispheric lesions consistent with focal cortical dysplasia (FCD): One was located in the posterior end of a sulcus in the right frontal lobe, anterior to the precentral sulcus, together with linear signal abnormality in the underlying white matter extending deep towards the right lateral ventricle (transmantle sign), and the second in the right parietal cortex, immediately posterior to the post-central gyrus (in the region of the supramarginal gyrus). Neurovascular contact with the trigeminal nerve had been excluded via an MRI study done previously. Interictal FDG-PET did not show any clear areas of hypometabolism.
(a) Axial T2w, coronal T2w and coronal FLAIR images showing two areas of subtle cortical thickening with subcortical high signal and blurring of the grey-white matter interface, one in the depth of a sulcus in the right frontal lobe (solid arrows) and another more posteriorly in the right parietal lobe involving the supramarginal gyrus (dashed arrows). Appearances of both lesions were consistent with FCD. (b) Corresponding post-surgical images following resection of the more anterior lesion showing a surgical cleft and subtle nonspecific signal changes along the margins/depth of the cavity. The more posterior lesion (dashed arrows) was not resected and remains unchanged.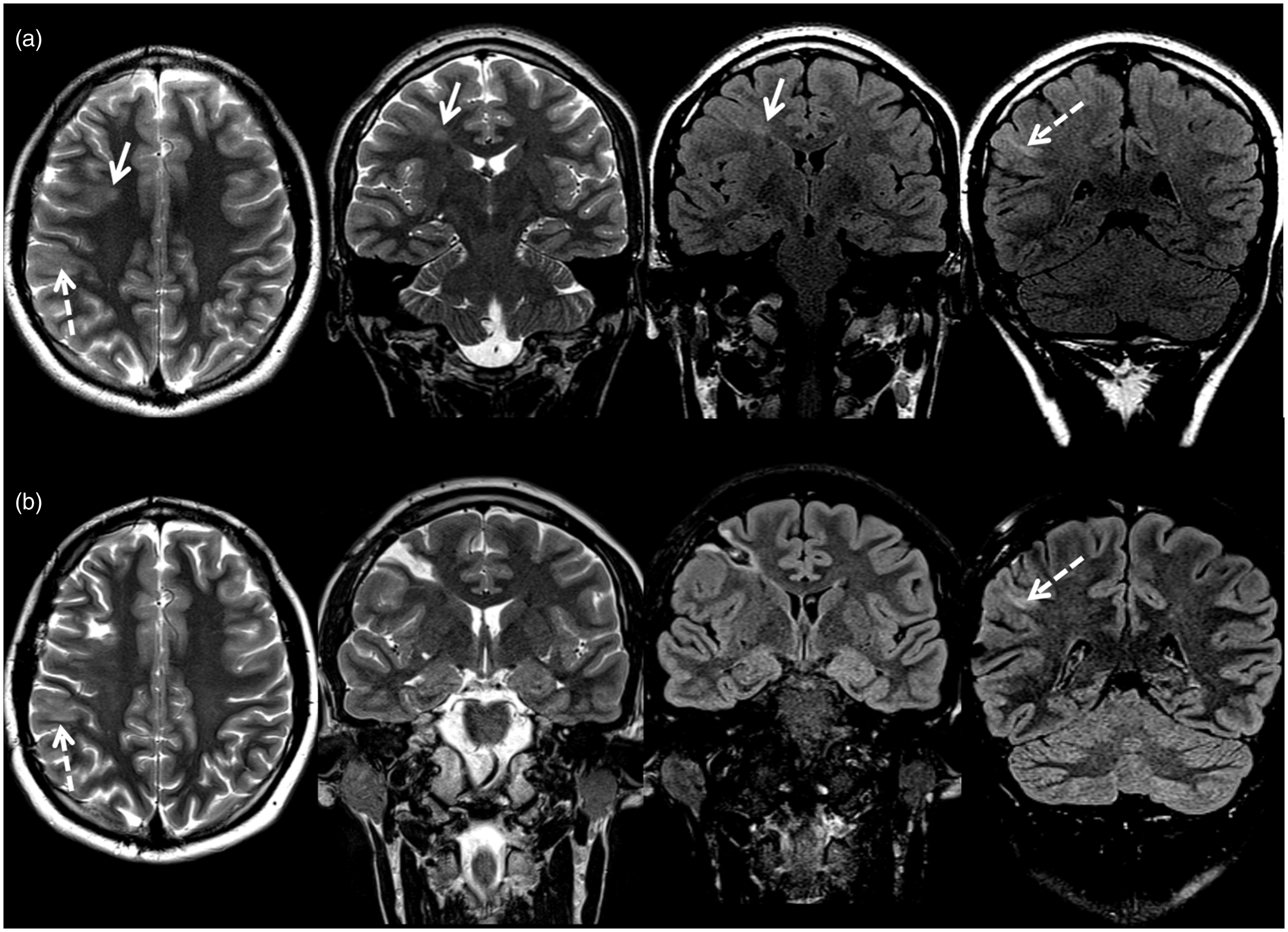
In order to establish which of the two lesions was causative, craniotomy with intraoperative electrocorticography (ECoG) was performed, with a plan to resect the lesion showing epileptiform discharges. However, ECoG using subdural electrodes showed no epileptiform discharges recordable intraoperatively. Therefore, free-hand depth electrodes around both lesions and two subdural 8-contact electrodes overlying the pre and post-central gyri were placed, for extraoperative recording. During subsequent telemetry, eight typical attacks were recorded arising from sleep. Ictal epileptiform discharges were recorded only from the subdural electrodes but were not clearly localised. However, electrical stimulation of the depth electrode adjacent to the frontal lesion consistently and immediately reproduced the patient’s habitual painful aura, time locked to the stimulation. Stimulation of the electrodes adjacent to the parietal lesion produced no responses. The frontal lesion was therefore resected. The post-operative MRI is shown in Figure 1(b). Histology confirmed a type IIB focal cortical dysplasia.
There was no post-operative neurological deficit, and the patient experienced immediate and sustained remission of his painful seizures, resulting in “the best night of sleep in the last 15 years”. He has remained seizure and pain-free for over two years and is currently off anti-epileptic medication. He has resumed his studies in aeronautical engineering.
Discussion
We describe a case of a refractory, short-lasting, unilateral retroorbital IEH, associated with a contralateral prefrontal lesion, cured by surgical resection. Free-hand implantation of the depth electrodes, resulting in suboptimal lesion targeting, may have limited the recording of ictal discharges from the depth electrode, but electrical stimulation from the electrode closest to the frontal lesion clearly reproduced the patient’s exact aura (not reproduced from the parietal electrodes), suggesting that the frontal lesion was the one responsible for the patient’s seizures, a finding corroborated by his long-term remission after resection.
Gowers first described pain as an epileptic manifestation, classifying painful seizures into hemicorporeal (unilateral), cephalic and abdominal (visceral or pneumogastric) (4).
Since then, several cases of headache as a prominent manifestation of seizures have been reported, and the first criteria for IEH were published in 2012 by Parisi et al. (5). IEH seems rare, reported in 0.1–0.7% in retrospective studies in epilepsy centres (6–8). It has been described in different epilepsy syndromes, including symptomatic focal epilepsy, generalised idiopathic epilepsy and occipital (Gastaut-type) epilepsy (9).
IEH cases are often difficult to diagnose. They require an ictal EEG recording during a headache attack, which by itself may not be conclusive (such as in our and other reported cases) (9). Simultaneous report of headache during ictal discharges and its remission after EEG offset is a challenge and is not well documented in many cases. Another feature for diagnosis of IEH (present in the 2012 criteria) is the electroclinical resolution after intravenous anti-epileptic medication, not possible in our case due to short seizure duration, rendering our diagnosis “Probable IEH”, according to the initially proposed criteria (9).
Different headache types have been described in IEH, the most frequent being migraine with or without aura and tension headache. However, there have been reports of IEH mimicking SUNCT (2), glossopharyngeal neuralgia (2) and trigeminal neuralgia (3).
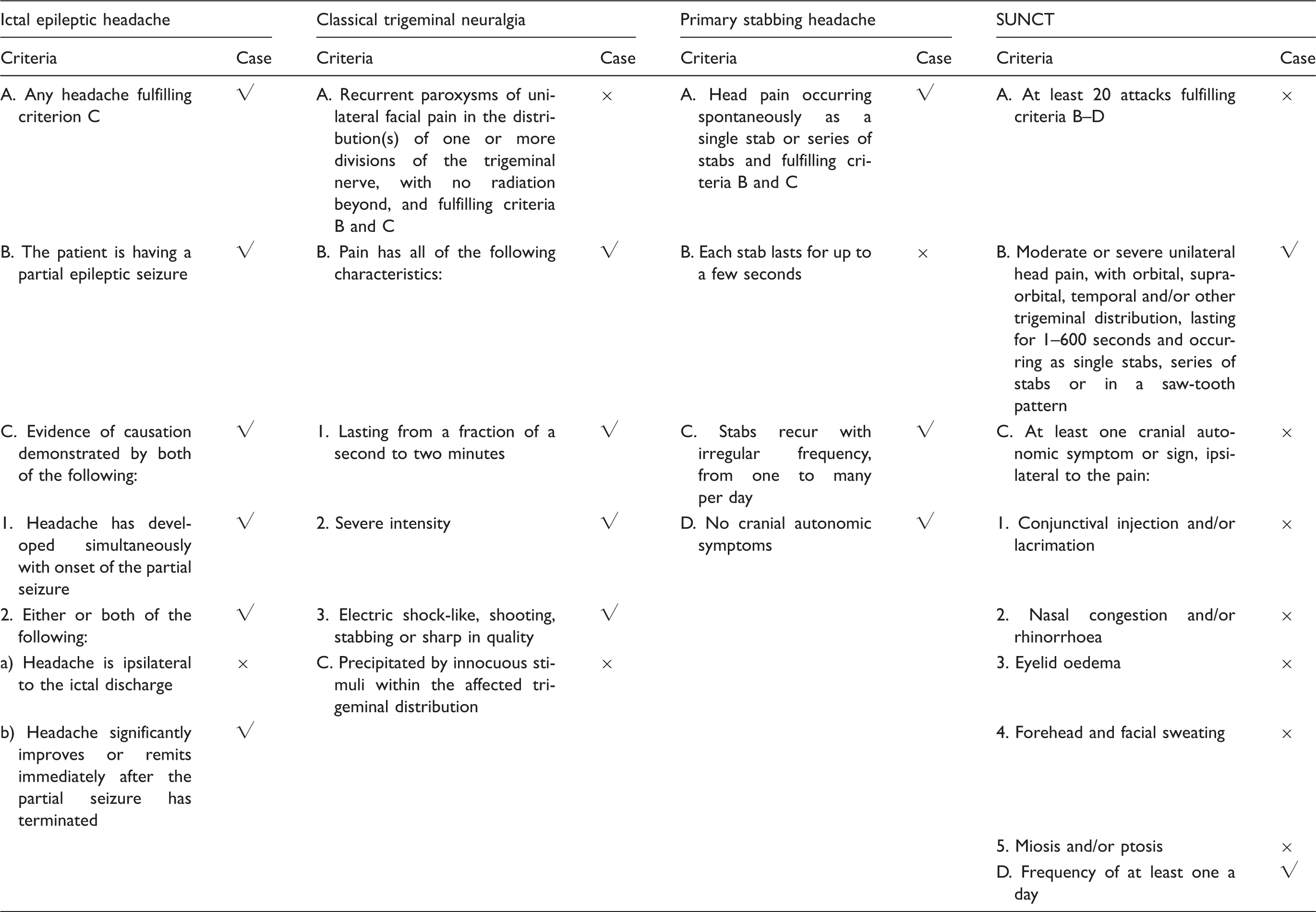
Comparison of our case with the diagnostic criteria for ictal epileptic headache and selected short-lasting primary headaches, according to the International Classification of Headache Disorders – third edition (1). Criteria “not better accounted by another ICHD-3 diagnosis” is not depicted.
Painful seizures were postulated to arise primarily from the insular (10) or the somatosensory parietal cortex (8). In our case, the epileptogenic and symptomatogenic networks involved the prefrontal cortex, in contrast with our a priori hypothesis that the parietal lesion might be epileptogenic.
Ictal cephalic sensations have been reported in a case series of premotor epilepsy (11). Other IEH cases also show anterior quadrant or frontotemporal ictal changes, although the majority were not intracranially explored (9).
The prefrontal cortex is a well-documented component of the pain matrix (12). Extremely well localized painful seizures, such as in ours and other cases, including with lesions outside classical somatotopical representation areas (2,3), are difficult to explain. One hypothesis involves the epileptic discharge exerting a direct modulatory effect of subcortical structures (hypothalamus or brainstem), a mechanism similarly postulated to occur in TACs such as cluster headache (12).
Another possibility is early spread to somatotopically represented cortex. Considering our case, stimulation of the parietal electrodes did not induce a typical aura. Furthermore, although propagation to the unsampled insula cannot be excluded, the absence of other well-described insular ictal symptoms (tachycardia, sudoresis, throat constriction, sense of suffocation and other autonomic symptoms) is against this hypothesis.
In conclusion, we describe a case of IEH caused by frontal lobe epilepsy, with long-term seizure and headache remission after resective surgery. IEH, albeit rare, should be considered in selected patients, particularly with clinical history of other seizure types, other ictal symptoms/signs, or neuroimaging abnormalities. A negative scalp ictal EEG recording does not exclude IEH. Finally, sampling the pre-frontal cortex should also be considered during neurophysiological exploration of painful epileptic seizures, and successful treatment might be achieved with resective surgery.
Clinical implications
Ictal epileptic headache (IEH) may mimic a variable number of classical headache syndromes, including short-lasting primary headaches. Resective surgery may constitute a treatment option in selected lesional IEH cases. Our case extends the neuroanatomical spectrum of IEH and reinforces the role of the prefrontal cortex in pain generation.
Footnotes
Acknowledgements
The reported patient provided full consent for publication of his clinical history and imaging.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.