
Abstract
Background
To further understand the role of pituitary adenylate cyclase-activating polypeptide 1 (PAC1) receptors in headache disorders, we mapped their expression in tissues of the trigemino-autonomic system by immunohistochemistry and in situ hybridization.
Methods
To optimize screening for monoclonal antibodies suitable for immunohistochemistry on formalin-fixed, paraffin-embedded tissues, we developed a new enzyme-linked immunosorbent assay using formalin-fixed, paraffin-embedded cells overexpressing human PAC1 receptors. 169G4.1 was selected from these studies for analysis of rat and human tissues and chimerized onto a mouse backbone to avoid human-on-human cross-reactivity. Immunoreactivity was compared to PAC1 receptor mRNA by in situ hybridization in both species.
Results
169G4.1 immunoreactivity delineated neuronal cell bodies in the sphenopalatine ganglion in both rat and human, whereas no staining was detected in the trigeminal ganglion. The spinal trigeminal nucleus in both species showed immunoreactivity as especially strong in the upper laminae with both cell bodies and neuropil being labelled. No immunoreactivity was seen in either rat or human dura mater vessels. In situ hybridization in both species revealed mRNA in sphenopalatine ganglion neurons and the spinal trigeminal nucleus, a weak signal in the trigeminal nucleus and no signal in dural vessels.
Conclusion
Taken together, these data support a role for PAC1 receptors in the trigemino-autonomic system as it relates to headache pathophysiology.
Keywords
Introduction
Clinical experiments have shown that pituitary adenylate cyclase-activating polypeptide 38 (PACAP-38, herein referred to as PACAP) elicits a migraine-like headache when administered systemically in migraineurs, thus implicating it in the pathophysiology of migraine (1,2). Plasma levels of PACAP are increased during migraine (3,4), and also cluster headache (5). Activation of the trigeminovascular system by PACAP has been proposed as the underlying mechanism causing headache (6,7). PACAP expression has been described in key relay structures of the trigeminovascular system; that is, the primary sensory neurons of the trigeminal ganglion (8–10) and the cell bodies and nerve fibers of the human spinal trigeminal nucleus (11–14). Furthermore, PACAP expression has been localized to fibers innervating the cerebral (15,16) and dural vasculature (10,16). PACAP expression is also prominent in the parasympathetic otic and sphenopalatine ganglia (also referred to as the pterygopalatine ganglia) that mediate trigemino-autonomic signaling (13,17). The mechanisms by which PACAP mediates its effects in the trigeminovascular system are less well understood. PACAP acts at three receptors: PAC1, VPAC1 and VPAC2. Several PAC1 receptor isoforms exist due to alternative splicing (18). PAC1 receptors have high affinity for PACAP and 100- to 1000-fold lower affinity for vasoactive intestinal peptide (VIP), whereas VPAC1 and VPAC2 receptors have equal binding affinity for PACAP and VIP (for reviews, see 19,20). Functional studies in rodent models of trigeminal sensitization and neurogenic dural vasodilation have implicated PAC1 receptors in mediating the effects of PACAP and suggest that targeting PAC1 receptors is a promising approach for migraine therapy (21,22). Understanding the localization of PAC1 receptors in the trigeminovascular and autonomic system is important to further unravel their potential role in migraine pathophysiology and to design potential therapeutics. Several studies have characterized PAC1 receptor mRNA and/or protein expression in humans and/or rodents: In intracranial arteries, mRNA expression of PAC1 receptors has been reported in rodents and humans (23,24) and immunoreactivity was demonstrated in rodents (25). In the sphenopalatine ganglia, PAC1 receptor mRNA could not be detected in either neurons or glia in human (26), but immunoreactivity was found in satellite glia and fibers, both in humans and rodents (13). Both PAC1 receptor mRNA and immunoreactivity were detected in the rodent and human trigeminal ganglion neurons (26,27) and PAC1 receptor mRNA was also reported in the rodent spinal trigeminal nucleus (28). Due to the use of different tools, tissues and species by the individual studies, some gaps in our understanding of the localization of PAC1 receptors remain. In the current study, we characterized the expression of PAC1 receptors in migraine-related tissues of rat and human using the same species cross-reactive antibody, 169G4.1. We then compared immunoreactivity with mRNA expression by in situ hybridization. To optimize detection of PAC1 receptors in formalin-fixed, paraffin embedded (FFPE) tissues, we screened monoclonal antibodies raised against the human PAC1 receptor extracellular domain using a novel enzyme-linked immunosorbent assay (ELISA) with FFPE cells overexpressing human PAC1 receptors. Antibodies that yielded a strong signal in this assay were further characterized on FFPE embedded cells overexpressing human and rat PAC1 receptors (both long and short isoforms) or VPAC2 receptors as control. Antibody 169G4.1 was selected for mapping PAC1 receptor expression in rat and human dura, sphenopalatine and trigeminal ganglia and the spinal trigeminal nucleus. PAC1 receptor staining was then compared to the in situ hybridization signal in these tissues using species-specific RNA probes.
Methods
PAC1 antibody generation and screening: Antibodies were generated by Amgen Research (Burnaby, CA). To generate PAC1 receptor-specific antibodies, the PAC1 extracellular domain 1 (GenBank accession no. AAQ72806.1 position 21–155) was used as immunogen. Xenomouse® animals were immunized using a standard procedure (29) and hybridomas were screened by fluorescence-activated cell sorting (FACS) for binding to the immunogen immobilized on beads. To identify antibodies suitable for FFPE tissues, approximately 400 antibodies that recognized the PAC1 immunogen in FACS were further screened by ELISA using FFPE cells overexpressing human PAC1 receptors as follows: CHO cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and stably transfected with human PAC1 receptors. Cells were cultured until approximately 80 million cells could be harvested. Cells were removed from tissue culture flasks with cell dissociation buffer (Invitrogen, Carlsbad, CA), washed in Dulbecco’s phosphate-buffered saline (PBS, Corning, Manassas, VA) and fixed in 10% neutral buffered formalin (NBF, Thermo Scientific, Waltham, MA) for 24 hours. Afterwards, cells were stored in 70% ethanol (EtOH) until embedded in paraffin in a Tissue-Tek VIP5 tissue processor (Sakura, Torrance, CA). The paraffin cell pellet was then de-paraffinized with xylene, four times, 4 minutes each, and re-hydrated through a descending EtOH series (two times, 3 minutes each of 100% EtOH, 95% EtOH, 70% EtOH,) and equilibrated in distilled water. Heat-mediated antigen retrieval was performed by incubating the cells in Diva Decloaker solution (Biocare, Concord, CA) in a Decloaking Chamber (Biocare, Concord, CA) at 125℃ for 30 seconds and 90℃ for 10 seconds. The Diva Decloaker solution was decanted from the cells and the cells were re-suspended at 1 × 105 cells/mL in Sniper blocking solution (Biocare, Concord, CA). Cells were added to a Nunc 96 well plate (Thermo Scientific, Waltham, MA) overnight at 4℃. The plates were spun at 1200 rpm for 5 minutes. Blocking solution was removed from the plate and cells were washed two times in ELISA wash buffer (ImmunoChemistry, Bloomington, MN). Plates were spun for 5 minutes at 1200 rpm between each wash. PAC1 receptor antibodies (Amgen Inc., Thousand Oaks, CA) were added at a concentration of 100 ng/mL and incubated for 1 hour at room temperature (RT). Antibodies were removed from the plate and cells were washed two times in ELISA wash buffer; plates were spun for 5 minutes at 1200 rpm between each wash. Mouse anti-human IgG (Jackson ImmunoResearch, West Grove, PA) was added for 30 minutes at RT followed by two washes in ELISA wash buffer; plates were spun for 5 min at 1200 rpm between each wash. Mouse Envision horseradish peroxidase (HRP) polymer (Dako, Camarillo, CA) was added for 30 minutes at RT and plates were again washed two times in ELISA wash buffer with a spin for 5 min at 1200 rpm between each wash. Finally, tetramethylbenzidine (TMB) substrate (Thermo Scientific, Waltham, MA) was added for 10 minutes at RT followed by stop solution (Thermo Scientific, Waltham, MA), and plates were read at an optical density of 450 nm. Antibodies with an optical density above 3.0 were then further analyzed by immunohistochemistry on FFPE cells as described below. Three antibodies including 169G4.1 were then cloned as a mouse IgG2a Fc chimeric, expressed in 293T HEK cells and purified antibodies were re-analyzed by immunohistochemistry on FFPE cells. 169G4.1 was chosen for tissue analysis based on selectivity and staining intensity. A mouse anti-human cadherin 19 antibody (Amgen Inc., Thousand Oaks, CA) was chosen as isotype control antibody in the tissues of interest.
169G4.1 selectivity testing and validation: CHO cells stably expressing human PAC1 receptors (Amgen Inc., Thousand Oaks, CA) or VPAC2 receptors (PerkinElmer, Waltham, MA) were grown in Ham’s F12, 10% fetal bovine serum (FBS), 1% glutamine/penicillin/streptomycin, 10 µg/mL Blasticidin, and 400 µg/mL G418. Stable rat PAC1 CHO cells were grown in Dulbecco’s modified essential medium (DMEM) with high glucose (Invitrogen, Carlsbad, CA) and 10% FBS, 1% hypoxanthine and thymidine (HT) supplement, 1% minimum essential medium (MEM) non-essential amino acids, 1% sodium pyruvate, 1% L-Glutamine, 10 µg/ml Puromycin dihydrochloride. For immunohistochemistry experiments, 5 × 107 cells were washed in PBS, pH 7.4, pelletized at 1200 rpm for 10 minutes; cell pellets were preserved in 10% NBF followed by paraffin embedding. Cell pellets were cut at 5 µm, mounted on charged microscope slides and stored at room temperature until processing for immunohistochemistry.
Rodent tissues: Three adult male Sprague Dawley® rats (6 – 8 weeks old) were obtained from Charles River. Rodents were kept in an AAALAC accredited facility and all procedures were performed in accordance with the U.S. Public Health Service Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Amgen Inc. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition (30). Animals were group-housed on corn cob bedding and had ad libitum access to food and water via an automatic watering system. Animals were maintained on a 12:12 hour light: dark cycle in rooms at constant temperature and humidity. Rodents were euthanized and dura, trigeminal and sphenopalatine ganglia and brainstem trigeminal nuclei were dissected, preserved in 10% neutral buffered formalin and embedded in paraffin.
List of human tissue samples.

Rodent tissue and cell pellet immunohistochemistry: Serial sections (4 µm) were cut from paraffin blocks, mounted on clean, charged microscope slides, and dried at 60℃ overnight. Paraffin was removed from the cell or rodent tissue sections with xylene and the sections were rehydrated with graded ethanol and immersed in distilled water. Antigen retrieval was performed using Diva Decloaker pretreatment reagent (Biocare Medical, Concord, CA) in a Biocare Decloaking Chamber (Biocare Medical, Concord, CA) set to reach 125℃ for 30 seconds, then 90℃ for 10 seconds. Tissue sections were processed at room temperature in an Intellipath FLX automated staining instrument (Biocare Medical, Concord, CA). Protein Block (Dako, North America) was applied to the tissue sections for 10 minutes. Tissue sections were incubated with mouse anti-PAC1 receptor antibody 169G4.1 (0.4 µg/ml, Amgen Corp. Thousand Oaks, CA) for one hour. Endogenous peroxidase was blocked using Hydrogen Peroxide Block (Biocare Medical, Concord, CA) for 10 minutes. Mouse-on-rat horseradish peroxidase (HRP) labelled Polymer (Biocare, Concord, CA) was used to detect the primary antibody. Sections were incubated with diaminobenzidine (DAB) plus chromogen substrate (Dako, North America) for 5 minutes. Sections were counterstained for one minute in hematoxylin (Richard Allan, San Diego, CA).
Human tissue immunohistochemistry: Serial sections were prepared at 4 µm and mounted onto charged microscope slides (Leica X-Tra™). Sections were de-paraffinized as described for the rodent tissues (see above), rinsed in PBS, pH 7.4, and then fixed in 10% NBF for 1 hour. Following fixation, sections were rinsed in PBS and equilibrated in distilled water for one minute each before loading into a ‘Decloaking Chamber NXGen’ antigen retrieval apparatus (Biocare Medical, Concord, CA). Heat-mediated antigen retrieval was performed by incubation of the sections for 10, 30 or 60 sec in Diva Decloaker solution (Biocare Medical, Concord, CA) at 110℃. Afterwards, slides were removed, washed in distilled water for 3 minutes and re-fixed in 10% NBF for 3 minutes. Slides were washed in PBS two times for 1 minute and loaded onto a Dako Autostainer. Sections were rinsed with Tris-buffered saline (TBS) and incubated for 15 minutes each with avidin blocker and then biotin blocker (Vector Laboratories, Burlingame, CA). Sections were incubated in two further blocking reagents, rodent blocker and FBS, each for 1 hour. Blocking agents were removed and sections were incubated with primary antibody, mouse anti-human PAC1 (Amgen 169G4.1, 0.4 µg/ml) in 50 mM Tris/1% bovine serum albumin for 1 hour. Following incubation with primary antibody, sections were rinsed three times in TBS and endogenous peroxidases were blocked by incubation with 3% H2O2/PBS for 1 hour. Sections were rinsed twice with TBS/ 0.1% Tween 20 and then incubated with Dako Envision anti-mouse HRP for 30 minutes followed by incubation in Dako DAB chromogen for 4 minutes. After two washes in TBS/0.1% Tween 20, slides were counterstained with haematoxylin, rinsed with deionized water, dehydrated in 100% ethanol, cleared with xylene and mounted under DePeX (Sigma, St. Louis, MO). Slides were dried in a fume hood for 24 h prior to digital scan.
Rodent tissue and cell pellet in situ hybridization: A 326 bp fragment from the 3’ end of the coding region corresponding to nucleotides 1448–1773 (Genbank #NM_133511) was cloned into the pCR4-TOPO plasmid vector (Invitrogen, Carlsbad, CA) and the template was verified by sequencing. The antisense 33P-labeled RNA probe was synthesized by in vitro transcription with T3 RNA polymerase (Promega, Madison, WI) after linearization with Not I restriction enzyme (New England BioLabs, Ipswich, MA). The sense 33P-labeled RNA probe was synthesized by in vitro transcription with T7 RNA polymerase (Promega, Madison, WI) after linearization with Spe I restriction enzyme (New England BioLabs, Ipswich, MA). In situ hybridization was performed in a humidified chamber overnight at 56℃ in hybridization solution with 10% dextran sulfate (Sigma-Aldrich, St. Louis, MO) and 1.5 × 106 cpm of 33P-labeled riboprobe per slide. After overnight hybridization, all slides were treated with RNase digestion (Sigma-Aldrich, St. Louis, MO; 20 µg/ml; 30 min; 37℃), followed by a series of 5 minute saline sodium citrate (SSC, Invitrogen, Carlsbad, CA) washes (2X SSC, 1X SSC, 0.5X SSC and 0.1X SSC) ending with the highest stringency of 0.1X SSC at 55℃ for 30 minutes. Slides were coated with NTB emulsion (Kodak, Rochester, NY) and exposed for 3 weeks in the dark at 4℃. Following exposure, slides were developed with D-19 Developer (Kodak, Rochester, NY), fixed in Professional Fixer (Kodak, Rochester, NY), counterstained with hematoxylin and eosin (American MasterTech, Lodi, CA), dehydrated through graded ethanol to xylene (Richard-Allan Scientific, San Diego, CA) and then cover slipped.
Human tissue in situ hybridization: The following four probe sets were applied in singleplex assay: PAC1 (NM_001118, probe VA1-20688), binding to residues 1855–2954, was purchased from Affymetrix (Santa Clara, CA). This probe hybridizes to all PAC1 transcripts. Beta-actin (βACT NM_001101 VA1-10351) was used as the positive control, Dihydrodipicolinate reductase (DapB) bacterial mRNA (L38424 VF1-11712) was used as the negative control. In addition, a probeset to insulin mRNA (INS NM_000207 VA1-10099) was applied to sections of human pancreas and served as the assay control (data not shown). For mRNA analysis using in situ hybridization, 12 µm frozen sections were prepared using a cryostat and mounted onto charged microscope slides (Leica X-Tra™, Richmond, VA). Sections were stored at −80℃ prior to use. Frozen sections were transferred from −80℃ to −20℃ (30 min) and then to +4℃ pre-chilled 4% formaldehyde in PBS, where tissue fixation occurred overnight (16–18 hours). Sections were washed in PBS (two times 1 min) and dehydrated through an ascending ethanol series: 50% EtOH, 70% EtOH, 100% EtOH, each for 10 min, and then baked at 60℃ ± 1℃ for 30 min. The dried slides were then loaded onto a LeicaBOND RX instrument for automated ISH processing using the ViewRNA™ ISH Tissue 1-Plex Assay Kit (ThermoFisher, Waltham, MA).
Image analysis: Slides were cleaned and either scanned using Aperio scanscope (Leica Biosystems, Richmond, VA) or visualized using a Zeiss microscope (Zeiss, Thornwood, NY). Images were assembled using Adobe Photoshop Elements 2.0 (Adobe Systems, San Jose, CA).
Results
169G4.1 was selected from a panel of approximately 400 anti-human PAC1 receptor antibodies that were screened using a novel FFPE ELISA assay (Figure 1(a)) and further analyzed by immunohistochemistry on FFPE cells overexpressing PAC1 receptors versus parental control cells. 169G4.1 stained cells expressing human PAC1 receptors, both long (Figure 1(b)) and short isoforms (Figure 1(c)). 169G4.1 also cross-reacted with rat PAC1 receptors (Figure 1(d)). No staining was detected on CHO parental cells (Figure 1(e)), cells overexpressing VPAC2 receptors (Figure 1(f)) or PAC1 overexpressing cells stained with an unrelated mouse-anti human IgG antibody (Figure 1(g)).
Screening and validation of mouse anti-human PAC1 receptor antibody 169G4.1 in cells overexpressing PAC1 receptors. Optical density of a panel of ∼400 PAC1 receptor antibodies was measured by FFPE ELISA (a). Each blue circle represents an antibody; 169G4.1 is marked in red. Immunoreactivity of mouse anti-human PAC1 antibody 169G4.1 was verified in FFPE embedded cells overexpressing the human PAC1 receptor long isoform (b), short isoform (c) or rat PAC1 receptors (d). Specificity was assessed by comparing to parental control cells (e), VPAC2 overexpressing cells (f) or PAC1 receptor overexpressing cells stained with a control IgG antibody (g). Scale bars = 50 µm.
In rat, no staining was detected in the trigeminal ganglion with either 169G4.1 (Figure 2(a)) or a control mouse anti-human IgG (Figure 2(b)). In contrast, PAC1 receptor immunoreactivity was detected in the sphenopalatine ganglion (Figure 2(c)) compared to no staining with the control antibody (Figure 2(d)). Neuronal cell bodies were lightly labelled, whereas strong immunoreactivity delineated the cells (Figure 2(c)). Strong PAC1 receptor immunoreactivity was also detected in the spinal trigeminal nucleus, particularly in the upper laminae (Figure 2(e) right hand, 2(f)) versus control (Figure 2(e) left hand). No immunoreactivity was detected in the rat dura with 169G4.1 (Figure 2(g)). The control antibody stained scattered cells in the dura, suggesting that the absence of staining with 169G4.1 was not due to tissue quality or the staining method used (Figure 2(h)).
Immunohistochemical analysis of mouse-anti human PAC1 receptor antibody 169G4.1 in rat tissues. No labelling of 169G4.1 was observed in the trigeminal ganglion (a) compared to control IgG (b). In contrast, strong immunoreactivity delineated a large portion of neuronal cell bodies in the sphenopalatine ganglion. Only faint intracellular labelling was seen in these neurons (c). No labelling was observed with the control antibody in the sphenopalatine ganglion (d). 169G4.1 immunoreactivity was distributed throughout the spinal trigeminal nucleus, but particularly strongly in the upper laminae ((e), right hand) compared to control antibody ((e), left hand). Both, fibers and neuronal cell bodies appeared to be labelled (f). Arrowheads indicate staining delineating a neuronal cell body (f). No immunoreactivity was observed in dura with 169G4.1 (g). The control antibody stained scattered cells in the dura indicating that the lack of immunoreactivity with 169G4.1 was not due to tissue quality or immunohistochemistry protocol (h).
In situ hybridization with a specific PAC1 receptor probe was used to compare the PAC1 receptor mRNA signal to the staining by 169G4.1 in rat tissues (Figure 3). In addition to a sense control (not shown), the specificity of the probe was validated in sections from CHO cells overexpressing PAC1 receptors (Figure 3(a)) compared to parental CHO cells (Figure 3(b)). In the trigeminal ganglion, few neurons were weakly labelled (Figure 3(c), (d)). In the sphenopalatine ganglion, strong labelling was seen in a large portion of the neurons (Figure 3(e), (f)). Labelling of the spinal trigeminal nucleus was seen throughout, but more densely in the upper laminae (Figure 3(g), (h)). No hybridization signal was detected in the blood vessels of the dura (Figure 3(k), (l)) or other CNS vasculature; for example, pial vessels and blood vessels in the circle of Willis (not shown).
In situ hybridization analysis of PAC1 receptor mRNA in rat tissues. Specificity of the rat PAC1 receptor probe was verified in sections from FFPE PAC1 receptor overexpressing cells (a) and compared to parental cells as control (b). Tissues were imaged in darkfield for maximal signal intensity ((a)-(c), (e), (g), (i), (k)) and brightfield with hematoxylin and eosin counterstain to visualize the tissue structures ((d), (f), (h), (j), (l)). In the trigeminal ganglion, low PAC1 receptor labeling was observed in scattered neurons ((c), (d)). In the sphenopalatine ganglion, most neurons displayed a prominent PAC1 receptor mRNA signal ((e), (f)). The spinal trigeminal nucleus showed an intense PAC1 receptor mRNA signal scattered throughout, with the highest concentration in the upper laminae ((g), (h)). No specific PAC1 receptor mRNA signal was detected in the meningeal vessels of the dura (asterisks, (i)–(l)). A brain section with adjacent dura vessel (asterisks) is shown in (k) and (l). Note the strong labelling in the adjacent brain tissue compared to background signal in the dural vessel ((k), (l)). Scale bars = 100 µm ((a)–(h)), 50 µm ((i)–(l)).
Staining of 169G4.1 in human tissue (Table 1) matched the staining pattern in rat. No staining was observed with 169G4.1 in neurons in any of the three human trigeminal ganglion samples analyzed. In one of the three samples, putative stained fibers were visible (Table 2, Figure 4(a)) . In contrast, strong immunoreactivity was found in both sphenopalatine ganglion samples (Table 2, Figure 4(c), (d)) compared to control (Figure 4(b)) IgG staining (Figure 4(e)). Neuronal cell bodies were lightly labelled, whereas strong immunoreactivity delineated the cells (Figure 4(c), (d), (f)). In the spinal trigeminal nucleus of three donors, labelling was found throughout and especially strongly in the upper laminae (Table 2, Figure 4(g) right hand, (h)). No staining was observed with control IgG (Figure 4(g) left hand). Neuronal cell bodies and fibers appeared to be labelled (Figure 4(i)). No staining was detectable in the human dura in three samples (Table 2, Figure 4(j)). The lack of immunoreactivity was not due to poor tissue quality, since prominent staining of the endothelial cells was seen with von Willebrandt factor in the same samples (Figure 4(k)).
Immunohistochemical analysis of mouse-anti human PAC1 receptor antibody 169G4.1 in human tissues. In the trigeminal ganglion ((a), (b)), no neuronal cell bodies were labelled by mouse anti-human PAC1 receptor antibody 169G4.1 (a) or control IgG (b). PAC1 receptor immunoreactivity was visible in fibers in one (a) out of three trigeminal ganglia from different donors. In the sphenopalatine ganglion ((c)–(f)) strong PAC1 receptor immunoreactivity was detected in all neurons ((c), (d), (f)) compared to IgG control (e). Cell bodies were only lightly labelled, whereas the strongest immunoreactivity was delineating the cells ((c), (d), (f)). A higher magnification of the sphenopalatine ganglion neurons in (d) is shown in (f). Note the strong labelling outlining the neurons. The spinal trigeminal nucleus ((g) right side, (i)) was labelled throughout with strongest immunoreactivity in the upper laminae ((g) right side, (h)). No labeling at all was visible with the control IgG ((g) left side). A 40× magnification of the upper laminae shows that both neuropil and cell bodies (arrowheads) express PAC1 receptor immunoreactivity (i) . No PAC1 receptor immunoreactivity was visible in meningeal arteries of the dura (j). Tissue quality was confirmed using von Willebrandt factor as the positive control antibody (k). Scale bars = 100 µm ((a)–(c), (i)–(k)), 50 µm ((d), (e)), 25 µm (f), 1.5 mm (g), 200 µm (h). Summary of staining results with 169G4.1 in human tissues.

The mRNA distribution pattern in human tissues was similar to the rat, although a different method with a human-specific probe set was used. All human tissues for in situ hybridization were obtained fresh frozen to optimize mRNA quality, with the exception of the sphenopalatine ganglia, where only FFPE tissue was available (Table 1). The specificity of the PAC1 in situ hybridization probe used on human tissues was confirmed on frozen sections from cells overexpressing PAC1 receptors and compared to parental cells with the same probe (not shown). In addition, actin RNA served as the positive (Figure 5 (b,e,h,k)) and DAPB mRNA as the negative control (Figure 5 (c,f, i, l)) for the PAC1 receptor mRNA signal. In the trigeminal ganglion, a weak PAC1 mRNA signal was detected in two of the four cases analyzed (Table 3, Figure 5(a)). Neurons in the sphenopalatine ganglion from two cases showed low to moderate levels of PAC1 mRNA (Table 3, Figure 5(d)) compared to high levels of actin mRNA (Figure 5(e)) and no signal with the DAPB control probe (Figure 5(f)). In the spinal trigeminal nucleus, a low PAC1 mRNA signal was detected in one of three cases analyzed (Table 3, Figure 5(g)) compared to a prominent actin mRNA signal (Figure 5(h)) and no signal with the control DAPB probe (Figure 5(i)). PAC1 mRNA was not detectable in dural arteries of any of the three cases analyzed (Table 3, Figure 5(j)). In contrast, actin mRNA yielded a strong signal in the smooth muscle cells of the same samples, suggesting that lack of PAC1 mRNA signal was not due to tissue quality (Figure 5(k)).
In situ hybridization analysis of PAC1 receptor, actin and DAPB mRNA distribution in human tissues. The hybridization signal of the PAC1 receptor probe ((a), (d), (g), (j)) was compared to the actin signal as positive control ((b), (e), (h), (k)) and DAPB signal as negative control ((c), (f), (i), (l)). In the trigeminal ganglion, a low PAC1 receptor mRNA signal was detected in a subset of neurons ((a), arrowheads) compared to a strong actin mRNA signal (b) and no signal with DAPB mRNA (c). In the sphenopalatine ganglion, PAC1 receptor mRNA was visible in all neurons (d) compared to controls ((e), (f)). In the spinal trigeminal nucleus, low amounts of PAC1 receptor mRNA were detected in neurons ((g), arrowheads) compared to prominent actin mRNA (h) and absence of DAPB mRNA (i). No PAC1 receptor mRNA was seen in dural vessels (j) compared to controls ((k), (l)). Scale bars = 50 µm ((a)–(i)), 100 µm ((j)–(l)). Summary of PAC1 receptor in situ hybridization in human tissues.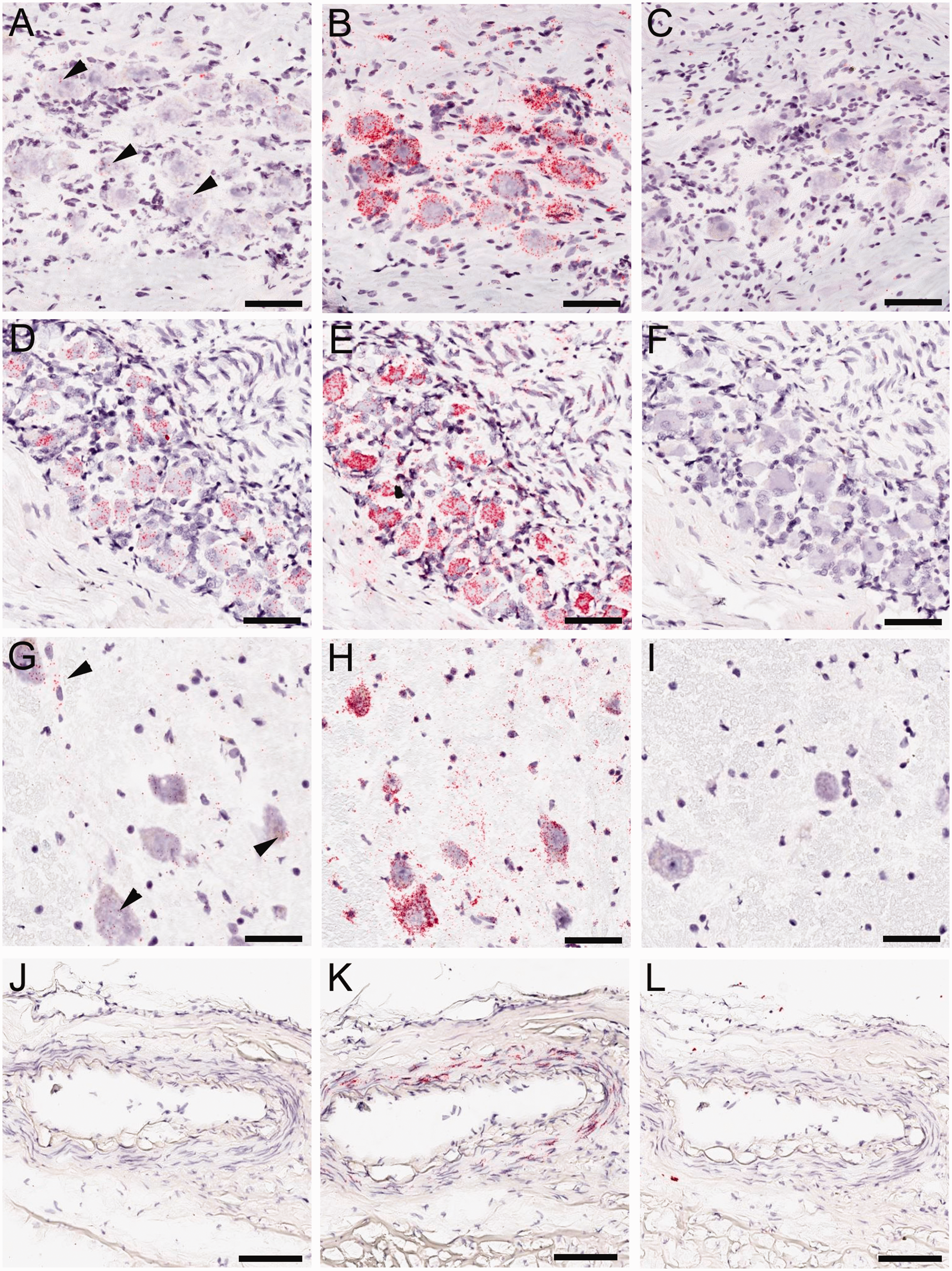

Discussion
In the current study, we compared immunoreactivity and mRNA expression of PAC1 receptors in rat and human trigeminal and sphenopalatine ganglion, spinal trigeminal nucleus and dural arteries. This is the first study that analyzed these four tissues for PAC1 expression in rat and human with the same cross-species reactive antibody, 169G4.1, which was discovered by FFPE ELISA screening of monoclonal antibodies generated against the extracellular domain of the human PAC1 receptor.
In the trigeminal ganglion, low levels of PAC1 mRNA expression were found in neurons in rat and two out of four human samples. The lack of mRNA in the other two samples may have resulted from stronger fixation and/or lower tissue quality due to postmortem interval. Unfortunately, we were not able to obtain information on these parameters for each donor tissue. In addition, the different causes of death of the tissue donors may have contributed to the variability in PAC1 mRNA expression observed. Published studies detected PAC1 mRNA in the rodent and human trigeminal ganglion neurons (26,27). Despite the presence of the low mRNA signal, no PAC1 immunoreactivity was detected in trigeminal ganglion neurons in either species. It is possible that while mRNA is located in the cell bodies, the actual PAC1 receptor protein is located on fibers travelling outside the trigeminal ganglion. Prominent PAC1 immunoreactivity was detected in the spinal trigeminal nucleus in both species and part of this could have been derived from afferent trigeminal fibers innervating the spinal trigeminal nucleus. Alternatively, PAC1 immunoreactivity may only be expressed under pathological conditions by the trigeminal ganglion neurons. PAC1 receptor up- or downregulation has been reported in several injury models or human diseases (31). One of three human cases showed fiber-like immunoreactivity in the trigeminal ganglion, but it was not possible to determine the origin of the fibers in the current study.
Sphenopalatine ganglia of both rat and human displayed PAC1 receptor mRNA and immunoreactivity in the majority of neurons. Immunoreactivity was faint or absent in neuronal cell bodies and concentrated at the cell membrane. It was difficult to distinguish whether some of the immunoreactivity was derived from surrounding satellite glia cells. The mRNA signal seemed to locate primarily to the neuronal cell bodies, and antibody 169G4.1 was raised against the extracellular domain of the PAC1 receptor that is expected to be outside the cell membrane. Therefore, it is likely that the immunoreactivity delineating sphenopalatine ganglion neurons represented neuronal membrane labelling rather than immunoreactivity from surrounding satellite glia. However, previous studies have localized PAC1 receptor immunoreactivity to satellite glia and fibers (13). Further studies using co-localization markers for glia cells are necessary to pinpoint the origin of the immunoreactivity in the sphenopalatine ganglion.
Rat spinal trigeminal nucleus showed PAC1 receptor mRNA concentrated in the upper laminae and scattered throughout the lower laminae, whereas a low level of mRNA level was detected in only one out of three samples of the human spinal trigeminal nucleus. As with the trigeminal ganglion, the lack of mRNA in two of the three spinal trigeminal nucleus samples could have been due to stronger fixation or lower tissue quality due to longer postmortem interval, or due to the different causes of death of the tissue donors. PAC1 receptor mRNA was previously reported by polymerase chain reaction in the rodent spinal trigeminal nucleus, although no functional PAC1 receptor responses were detected in the same tissue in that study (28). Prominent staining was found in the spinal trigeminal nucleus in both rat and human despite the low mRNA signal in the latter. Since the frozen human tissues used for in situ hybridization were from different donors than those used for immunohistochemistry, we cannot make a direct visual correlation of mRNA signal and staining intensity in the current study. Immunoreactivity was particularly strong in the upper laminae of the spinal trigeminal nucleus. Notably, PACAP immunoreactivity has been shown to exhibit a similar distribution pattern in this structure (14).
Neither rat nor human dural vessels showed PAC1 receptor mRNA expression or staining by 169G4.1. In contrast to our findings, mRNA expression of PAC1 receptors has previously been reported in rodent and human dural vessels (23,24) and immunoreactivity was demonstrated in rodents (25). Previous reports have used polymerase chain reaction to amplify the mRNA from dural vessels and could have derived the signal from innervating fibers rather than the smooth muscle cells themselves (23,24). In addition, the lack of immunoreactivity in the current study could be due to differences in the antibodies used between our study and published studies (25).
Taken together, our data show expression of PAC1 in the central trigemino-autonomic pathway in both rodents and humans. Peripherally, strong expression in both species was found in the sphenopalatine, but not trigeminal ganglion or dural blood vessels. These findings suggest that PACAP does not mediate dural vasodilation directly by acting on PAC1 receptors. Supporting this hypothesis, in a rat model of trigeminal sensitization the effects of PACAP on spontaneous firing of trigeminovascular neurons were found to be mediated by central PAC1 receptors, whereas peripheral vasodilation by PACAP was mediated by action on VPAC2 receptors (6). Peripheral actions of PACAP via PAC1 receptors may be mediated mainly by the sphenopalatine ganglion, where most neurons expressed PAC1 receptors on the cell membrane. The sphenopalatine ganglion is a large extracranial parasympathetic ganglion with viscerosensory and visceromotor roots (32). The parasympathetic inputs to the sphenopalatine ganglion derive from the superior salivatory nucleus in the brainstem, which in turn is activated by trigeminal inputs (33). Efferent parasympathetic fibers of the sphenopalatine ganglion innervate the lacrimal, nasal, palatine and pharyngeal glands (34) and the dural and cerebral blood vessels (35). Stimulation of the sphenopalatine ganglion has been shown to activate cerebral vasodilatation and increase cerebral blood flow by triggering the release of acetylcholine, vasoactive intestinal peptide and nitric oxide (36). This may increase plasma protein extravasation with resultant neurogenic inflammation and activation of trigeminal nociceptors contributing to pain and triggering headache (37). Therapies blocking the sphenopalatine ganglion have shown efficacy in headache disorders, particularly cluster headache, and are less robust in migraines (38). Inhibiting PAC1 receptor-mediated activation of central trigeminal transmission and/or parasympathetic output from the sphenopalatine ganglion may represent a novel therapeutic strategy for headache disorders. It was recently shown that the novel anti-CGRP antibody drugs block Aδ-fiber mediated signaling to high-threshold neurons in the spinal trigeminal nucleus, but not C-fiber mediated signaling to wide-range neurons in rats (39,40). Future studies investigating the functional role of PAC1 receptors in this model may reveal whether interfering with PAC1 receptors could silence additional populations of neurons involved in trigeminal nociceptive transmission.
Key findings
PAC1 receptor protein and mRNA expression in the spinal trigeminal nucleus, dura mater, trigeminal and sphenopalatine ganglion is similar in rat and human. PAC1 receptor protein and mRNA are expressed in rat and human sphenopalatine ganglion and spinal trigeminal nucleus, but could not be detected in rat or human dura mater and the trigeminal ganglion. This expression pattern suggests a role for PAC1 receptors in trigemino-autonomic signaling.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Kelly Hensley, Jim Pretorius, Brian Chan, Hantao Liu, Chang Choi, Di Shi, Cen Xu and Silke Miller were Amgen employees at the time of the study. Lars Edvinsson has provided consulting services to Amgen.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Amgen paid for all materials and services in this study including the human tissue staining and in situ hybridization performed by Keith Page at Asterand.