
Abstract
Objective
To study the position of hemiplegic migraine in the clinical spectrum of migraine with aura and to reveal the importance of CACNA1A, ATP1A2 and SCN1A in the development of hemiplegic migraine in Finnish migraine families.
Methods
The International Classification of Headache Disorders 3rd edition criteria were used to determine clinical characteristics and occurrence of hemiplegic migraine, based on detailed questionnaires, in a Finnish migraine family collection consisting of 9087 subjects. Involvement of CACNA1A, ATP1A2 and SCN1A was studied using whole exome sequencing data from 293 patients with hemiplegic migraine.
Results
Overall, hemiplegic migraine patients reported clinically more severe headache and aura episodes than non-hemiplegic migraine with aura patients. We identified two mutations, c.1816G>A (p.Ala606Thr) and c.1148G>A (p.Arg383His), in ATP1A2 and one mutation, c.1994C>T (p.Thr665Met) in CACNA1A.
Conclusions
The results highlight hemiplegic migraine as a clinically and genetically heterogeneous disease. Hemiplegic migraine patients do not form a clearly separate group with distinct symptoms, but rather have an extreme phenotype in the migraine with aura continuum. We have shown that mutations in CACNA1A, ATP1A2 and SCN1A are not the major cause of the disease in Finnish hemiplegic migraine patients, suggesting that there are additional genetic factors contributing to the phenotype.
Keywords
Introduction
Hemiplegic migraine (HM) is a rare and severe subtype of migraine with aura. It is characterized by fully reversible and unilateral motor weakness (hemiparesis), but otherwise it has similar clinical features as migraine with aura (1). Familial hemiplegic migraine (FHM) is diagnosed when there is at least one first- or second-degree relative in the family with HM, otherwise sporadic hemiplegic migraine (SHM) is diagnosed, while both share similar clinical characteristics (1,2). Severe attacks may occur in both forms with prolonged hemiplegia, confusion, coma, fever, and CSF pleocytosis. The clinical spectrum may also include permanent cerebellar signs (nystagmus, ataxia, dysarthria) and less frequently epilepsy (1).
FHM has been suggested to be a monogenic, genetically heterogeneous channelopathy disorder, with three definite causative genes, CACNA1A (FHM1) (3), ATP1A2 (FHM2) (4), and SCN1A (FHM3) (5). Despite their role in individual families with several affected family members, mutations in these three genes do not seem to be the major cause of HM in the general population (6,7), suggesting additional causal or contributing variants/genes for HM. A few other genes have been suggested, but their causality has not been confirmed (8).
This study is part of the Finnish Migraine Genome Project, a large ongoing family study focusing on clinical and genetic aspects of migraine. Here, we investigate the clinical characteristics and occurrence of HM in Finnish migraine families and study the involvement of CACNA1A, ATP1A2 and SCN1A in HM. Furthermore, we compare the clinical and genetic characteristics of this questionnaire-based family sample to previous population and clinical-based studies on hemiplegic migraine.
Material and methods
Patients
All participants gave informed consent, and approval to conduct the research was obtained from the Ethics Committee of the Hospital District of Helsinki and Uusimaa (111/13/03/01/2011, 3.8.2011).
The Finnish Migraine Genome Project is a large ongoing family study. It was initiated in 1992 focusing, at first, on migraine with aura families. Migraine patients and their relatives are recruited from headache clinics around Finland (primarily from Helsinki, Turku, Tampere, Jyväskylä, Kuopio and Kemi) and the Finnish Migraine Association. The participants are asked to fill out the validated Finnish Migraine-Specific Questionnaire for Family Studies (9) and to donate a blood sample for genetic studies. In addition, the index patients are clinically examined. Genomic DNA is extracted from peripheral blood cells with standard protocols.
By July 2014, 9087 migraine patients and their family members had taken part in the study (Figure 1). Altogether, 3383 subjects fulfilling the criteria for migraine with aura were included in this study. Of these, 406 patients had experienced two or more migraine attacks with fully reversible motor weakness and thus formed the hemiplegic migraine group (HM-group). The rest of the patients (n = 2977) formed the non-hemiplegic migraine with aura group (NHMwA-group) (Figure 1). All diagnoses were based on the criteria of the International Classification of Headache Disorders, 3rd edition, beta version (ICHD-3 beta criteria) (1).
Flowchart of the clinical and genetic analyses. MwA: migraine with aura; HM: hemiplegic migraine; FHM: familial hemiplegic migraine; SHM: sporadic hemiplegic migraine; NHMwA: non-hemiplegic migraine with aura; MAF: minor allele frequency; gnomAD: The Genome Aggregation Database; SISu: The Sequencing Initiative Suomi Database; VEP: the Variant Effect Predictor; SIFT (Sorts Intolerant From Tolerant) and PolyPhen2 (Polymorphism Phenotyping v2): variant prioritization tools.
According to the ICHD-3 beta, 131 patients were diagnosed with FHM and 275 patients with SHM. SHM patients had a diverse familial background for migraine, including third-degree or more distant relatives with HM and closer relatives with other subtypes of migraine. Altogether 24% of the SHM families had more than five family members with migraine with or without aura in addition to the SHM cases (Figure 2). Seventy-two patients with HM were classified as SHM due to unknown family history for migraine.
Migraine load in the studied families. SHM: sporadic hemiplegic migraine; FHM: familial hemiplegic migraine.
The 131 FHM patients belonged to 51 families. Most families had two (n = 32) or three (n = 12) FHM patients, while there were seven families with more than three FHM patients. Most of the families (94%) also contained members diagnosed with migraine with aura and/or migraine without aura (Figure 2).
Statistical methods
The HM and NHMwA groups were compared for 59 clinical variables related to migraine headache, migraine aura and migraine comorbidities. Within the study, all anamnestic data of seizures (also febrile seizures), any epileptic activity and sudden loss of consciousness, were classified as paroxysmal symptoms.
Statistical analyses were performed using IBM SPSS Statistics 22.0.0.0. In order to determine association of nominal variables across patient groups, Fisher's Exact test was used for 2 × 2 tables, while Pearson's Chi-Square test was used for larger tables. The Mann-Whitney U test was used for comparing means. Missing data were excluded from the analyses (see Supplementary tables 1–3 for response rate details). All reported p-values are two-sided and not corrected for multiple testing. Results with p < 0.05 were considered as evidence of significant differences between the groups, though the plausibility of type I errors should be kept in mind while interpreting the results.
We applied explorative principal components analysis (PCA) (10) to the clinical variables listed in Figure 3, Figure 4 and Figure 5, excluding the HM-specific symptoms (four related to motor weakness and two to clumsiness) from this analysis. All inspected 53 phenotypes were coded into 75 dichotomous dummy variables. After removing 653 individuals (24 HM cases) having 50% or more missing answers, the remaining missing answers were interpreted as no-answers. The final PCA data consisted of 2730 individuals (382 HM cases). To identify the number of relevant PCA components, we used Horn's parallel analysis (10), which compares eigenvalues from the observed data with eigenvalues of simulated data of the same size.
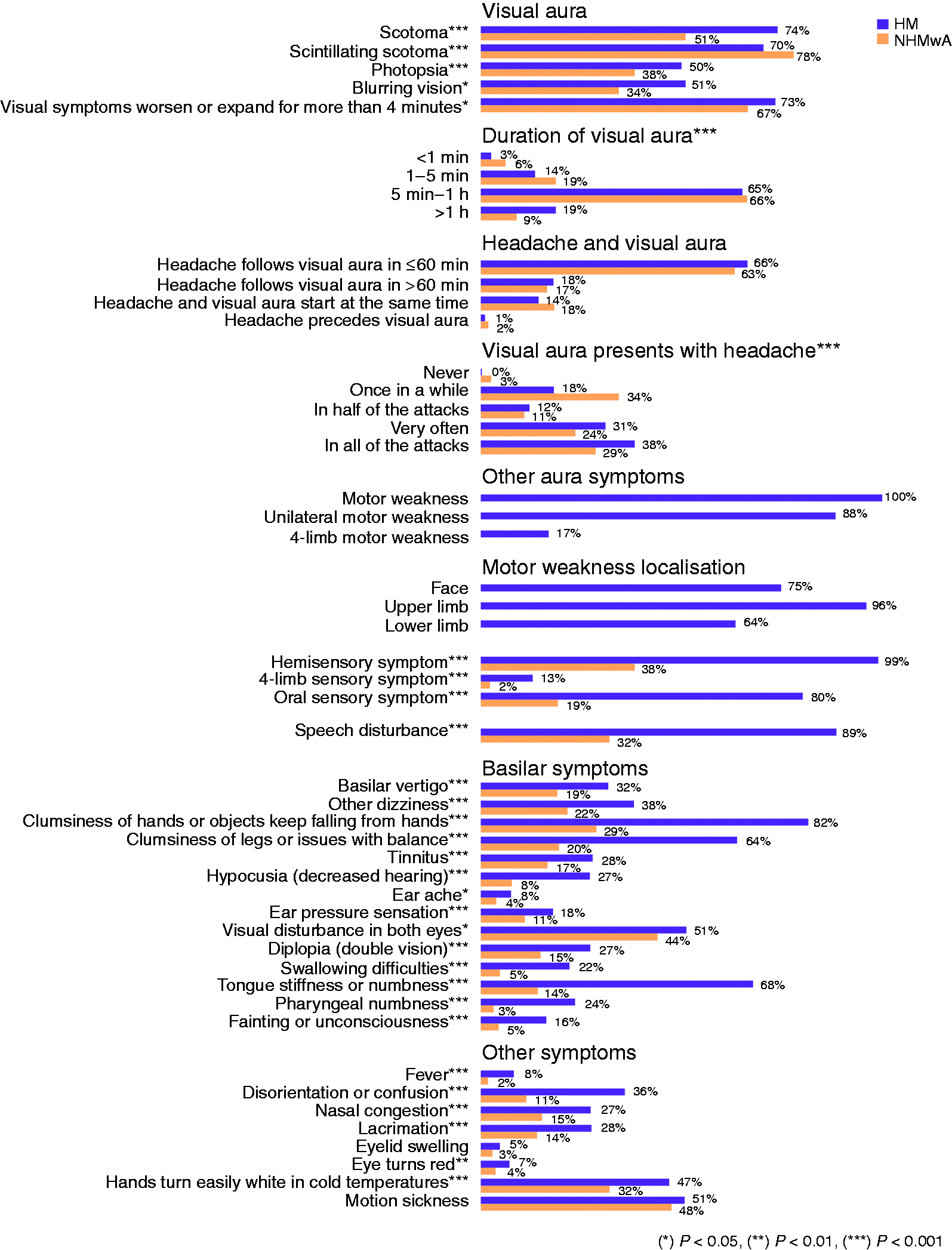
Characteristics of headache in hemiplegic migraine (HM) patients compared to non-hemiplegic migraine with aura (NHMwA) patients. Characteristics of aura in hemiplegic migraine (HM) patients compared to non-hemiplegic migraine with aura (NHMwA) patients. Comorbidities in patients with hemiplegic migraine (HM) compared to non-hemiplegic migraine with aura (NHMwA) patients.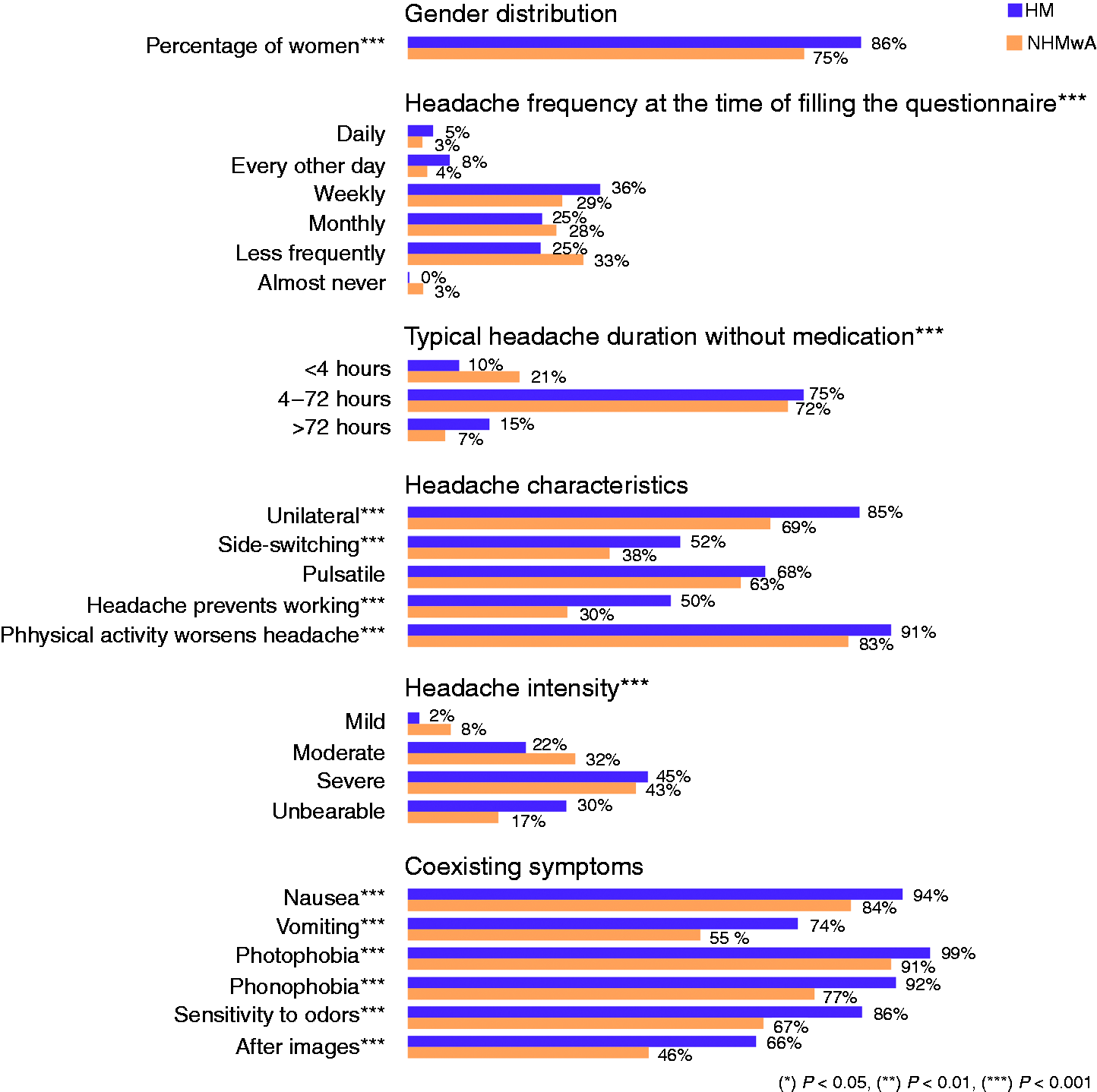

We illustrated the correlation structure of individual migraine characteristics using polychoric correlations (10). Polychoric correlations are a common way to assess correlations in self-report instruments such as health questionnaires having items with a small number of response options. To further group related symptoms, hierarchical clustering was conducted using Ward's agglomerative method (11). It is based on sum-of-squares criterion and produces groups by progressively merging similar observations together.
Whole exome sequencing (WES)
Sample selection
From the 406 HM patients, 303 DNA samples were sent for whole exome sequencing (WES). Individuals were selected based on a) their neurological motor symptoms (hemiplegia or hemiparesis) and b) the family structure. From each FHM family, the clinically examined index patient and one to three additional affected relatives were chosen. Of the 51 FHM families included in the clinical analysis, there are seven families from which we do not have WES data. These include one large FHM family with a previously identified mutation (p.Thr345Ala) in the ATP1A2 gene (12) that was intentionally excluded from the WES analysis, five families from which no DNA samples were available, and one family from which the sequencing failed. Thus, sequencing results were successfully obtained from 293 subjects (92 FHM and 201 SHM patients) from 243 families (44 FHM and 199 SHM) (Figure 1).
Exome sequencing
WES was performed at the Wellcome Trust Sanger Institute (Hinxton, Cambridge, UK) throughout the years 2010–2013, using the SureSelect Human All Exon 50 Mb capture protocol (Agilent Technologies, Santa Clara, CA, USA) and the HiSeq next-generation sequencing system (Illumina, San Diego, CA, US). The sequence data were aligned to the human reference genome, NCBI Build 37 (GRCh37/hg19; The Genome Reference Consortium), and variants were jointly called and filtered together with 10142 Finnish exomes as part of the Sequencing Initiative Suomi (SISu) project (13). Variant calling was performed at the Broad Institute of Harvard and MIT, and quality control at the Institute for Molecular Medicine Finland (FIMM).
Variant filtering
We performed variant analysis for variants in CACNA1A, ATP1A2 and SCN1A (Figure 1). The average sequencing depth (DP) across the genes was 26 for CACNA1A, 33 for ATP1A2 and 36 for SCN1A. Variants were filtered in a step-wise manner to identify alterations that were rare (minor allele frequency < 0.01) in the Genome Aggregation Database (gnomAD) (14) and functionally likely to be damaging (missense, splicing, stop gain, stop loss, in-frame insertion, in-frame deletions or frameshift) based on the Variant Effect Predictor (VEP) annotations (Ensembl Release 75) (15). Annotations were based on the Ensembl database's canonical transcripts: ENST00000 360228.5 (CACNA1A), ENST00000361216.3 (ATP1A2) and ENST00000303395.4 (SCN1A).
In the second filtering step (Figure 1), we considered only variants that are very rare or absent in the Finnish population (MAF < 0.001 in the SISu database) to be potential causal mutations. Furthermore, to interpret the causality, we utilized SIFT (Sorts Intolerant From Tolerant) (16) and PolyPhen2 (Polymorphism Phenotyping v2) (17) prediction algorithms, paralog conservation score calculations (18), co-segregation analyses and previously published literature. The SIFT and PolyPhen2 tools were used to predict the pathogenicity of missense variants and paralog conservation scores (para_zscore) to evaluate conservation of the amino acid within the gene family. Missense variants that have positive paralog conservation scores (para_zscore > 0), resulting in changes in highly conserved amino acid residues, are more likely to be deleterious.
Validation
Validation of candidate variants and studying co-segregation in family members were done by Sanger sequencing (FIMM Sequencing Unit, Helsinki, Finland). Primers were designed with Primer-BLAST (19) and sequences were analyzed using Sequencher (Gene Codes Corporation, Ann Arbor, MI, US). Primer sequences and PCR conditions are available from the authors on request.
Control allele frequency databases
Validated variants of ATP1A2, SCN1A and CACNA1A identified based on the exome sequencing data on hemiplegic migraine (HM) patients. The variants considered to be causal are highlighted with orange and one probably causal variant with purple background color.
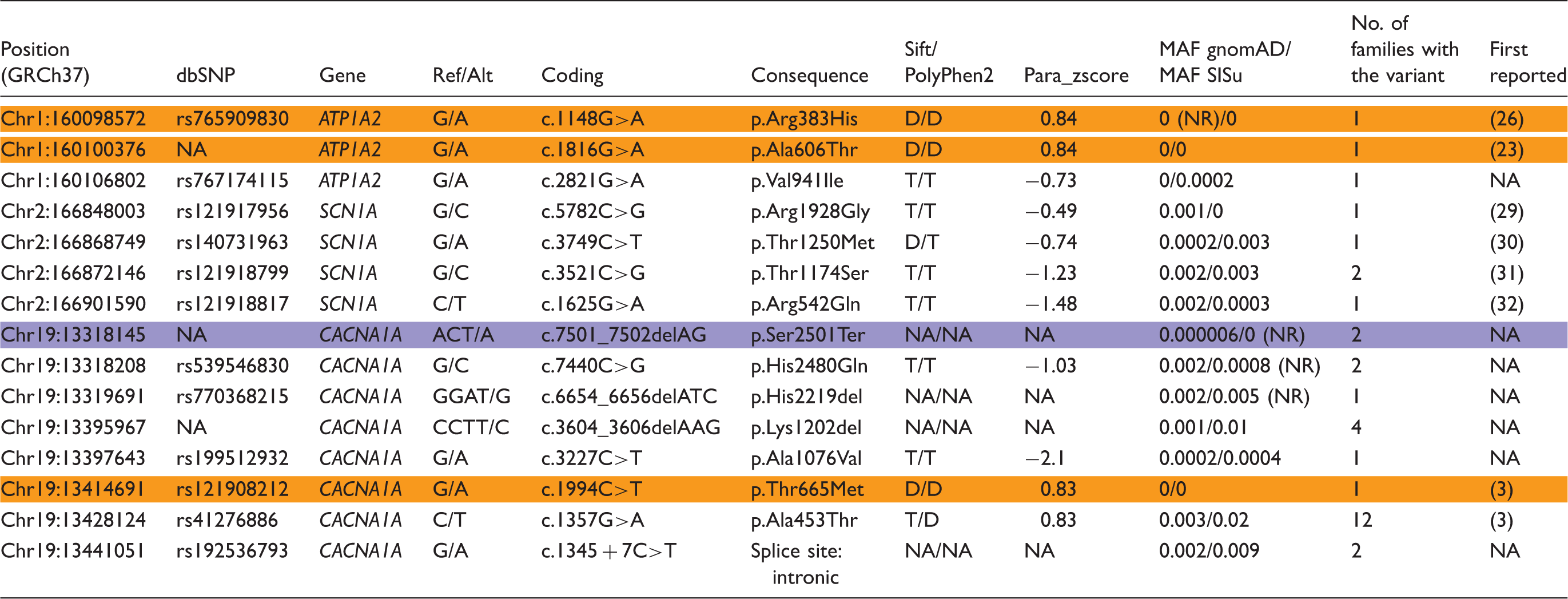
Sift/PolyPhen2: Variant pathogenicity prediction tools; D: deleterious/probably damaging; T: tolerated/benign; Para_zscore: paralog conservation score, positive values indicate conservation of the amino acid within the gene family; MAF gnomAD: minor allele frequency in The Genome Aggregation Database (gnomAD; Finnish data excluded); MAF SISu (The Sequencing Initiative Suomi): minor allele frequency in the Finnish population (our migraine cohort excluded); NA: not applicable; NR: not reliable – the variant site is covered in fewer than 80% of the individuals in gnomAD or SISu, thus the allele frequency cannot be reliably estimated; reference sequences: ENST00000361216.3 (ATP1A2), ENST00000303395.4 (SCN1A) and ENST00000360228.5 (CACNA1A).
Results
Clinical characteristics
Clinical characteristics of the 406 studied HM patients are described in Figure 3, Figure 4, and Figure 5. HM patients were younger (42.6, SD ± 14.0) than NHMwA patients (47.6, SD ± 16.7, p < 0.001) and a higher proportion of them were women (85.7% vs. 75.0%, p < 0.001). Furthermore, HM patients reported earlier age at onset of headache, with median of 12 years compared to NHMwA patients with median of 14 years (IQR 9–17 and 10–20 years respectively; p < 0.001, response rate for HM 84.7% and NHMwA 67.8%).
According to ICHD-3 beta criteria, HM patients reported more severe headache characteristics compared to NHMwA patients (Figure 3). For example, at the time of filling the questionnaire, they reported having daily or almost daily headache more often than NHMwA patients (12.7 vs. 6.5%, p < 0.001). Furthermore, they had unbearable headache (30.0 vs. 17.1%), work-preventing headache (49.7 vs. 30.2%), and headaches lasting over 72 hours (15.4 vs. 7.0%) more often than NHMwA patients (p < 0.001). HM patients also experienced nausea and vomiting more often, as well as light, smell and sound sensitivity compared to NHMwA patients (p < 0.001). The only headache characteristic that was not significantly different between the patients' groups was the prevalence of pulsation (Figure 3).
In addition to headache characteristics, aura episodes were also more severe in HM patients compared to NHMwA patients (Figure 4). Almost all patients reported visual disturbances, but a higher proportion of attacks involved visual symptoms in HM patients than in NHMwA patients (68.2 vs. 52.7%, p < 0.001). Visual aura lasting over an hour (18.7 vs. 8.9%), hemisensory symptoms (99.0 vs. 38.3%, p < 0.001) and speech disturbances (88.6 vs. 32.1%, p < 0.001) were also more prevalent in HM patients than in NHMwA patients. Furthermore, basilar-type aura symptoms including bilateral sensory symptoms, basilar vertigo, tinnitus, hypoacusia, diplopia, swallowing difficulties, unconsciousness, and confusion were more often reported by HM patients than NHMwA patients (p < 0.001) (Figure 4). Almost all evaluated non-migraine specific coexisting symptoms were similarly more common in HM patients than in NHMwA patients, the only exceptions being motion sickness and the swelling of eyelids (Figure 4).
In addition to other clinical characteristics, HM patients reported more psychological disorders (23.9 vs. 17.7%, p = 0.005), allergy (45.0 vs. 34.5%, p < 0.001), asthma (12.7 vs. 9.4%, p = 0.049), and Raynaud's phenomenon (21.9 vs. 13.9%, p = 0.001) than NHMwA patients. There were no significant differences in the prevalence of stroke, epilepsy, or diabetes (Figure 5). The reported prevalences are not adjusted for age.
Visualisation of migraine symptom spectrum using principal component analysis
To collectively visualize diverse migraine symptoms and examine potential clustering of patients and traits, we applied explorative PCA analysis to the phenotype data. Eigenvalue distribution suggests retaining only the first principal component that alone explains 49.8% of the variance among all inspected symptoms. However, to assess any potential clustering, we further visually investigated first 10 components that together explain 67.6% of all variance. None of these 10 components seem to separate individuals into distinguishable or pronounced groups. Rather, the distribution seems to be continuous across all inspected components. For illustration, the first two principal components of 53 migraine symptom characteristics across 2730 individuals are plotted in Figure 6, highlighting the variant carriers from Table 2. Most HM patients seem to concentrate to the other end of the first component, which explains nearly half of the overall symptom diversity (Figure 6). Component loadings for these first two components can be found in Supplementary Figure 1. The correlation structure of individual symptoms is illustrated in Supplementary Figures 2 and 3.
First two principal components of 53 migraine symptom characteristics across 2730 individuals. Hemiplegic migraine (HM) patients seem to be at the other end of the spectrum but do not form a separate, bounded cluster. Variant carriers from four families (A,B,C,D) are highlighted with squares. Family trees of A, B and C are presented in Figure 7. Family D is a large familial hemiplegic migraine family with a previously identified mutation (p.Thr345Ala) in ATP1A2 gene (12). Clinical features of the family members with CACNA1A and ATP1A2 mutations. F: Female; M: Male; Age: the participation age; AO: age at onset of migraine; NA: not applicable; + : symptom present in all or some attacks, −: symptom never present; FHM1: familial hemiplegic migraine type 1; FHM2: familial hemiplegic migraine type 2; MwA: migraine with aura; MwoA: migraine without aura; NoHa: no headache.

Genetic findings
To study the involvement of CACNA1A, ATP1A2 and SCN1A in the HM phenotype, we exome sequenced samples of 293 patients with HM from 243 Finnish migraine families. We identified and validated 15 potentially damaging, rare frequency variants (Figure 1, Table 1). Eleven of the validated variants did not completely fulfill our predetermined mutation criteria, whereas c.1994C > T (p.Thr665Met) in CACNA1A, c.1148G > A (p.Arg383His) and c.1816G>A (p.Ala606Thr) in ATP1A2 were considered as causative variants. Their pathogenicity is further supported by their location in highly conserved amino acids and previous clinical and functional studies. In addition to the three pathogenic missense variants, we identified one probable causative frameshift variant c.7501_7502delAG (p.Ser2501Ter) in CACNA1A.
The p.Thr665Met likely causal mutation in CACNA1A was found in one family where it co-segregates with the HM phenotype. However, the proband's elder sister, who does not have migraine, carries the mutation, leading to a penetrance of 75% (Figure 7, family A, II:2).
Pedigrees of the Finnish familial hemiplegic migraine families with mutations in the CACNA1A and ATP1A2 genes. Mutation is indicated with + and wild type allele with −; reference sequences: ENST00000361216.3 (ATP1A2), ENST00000360228.5 (CACNA1A).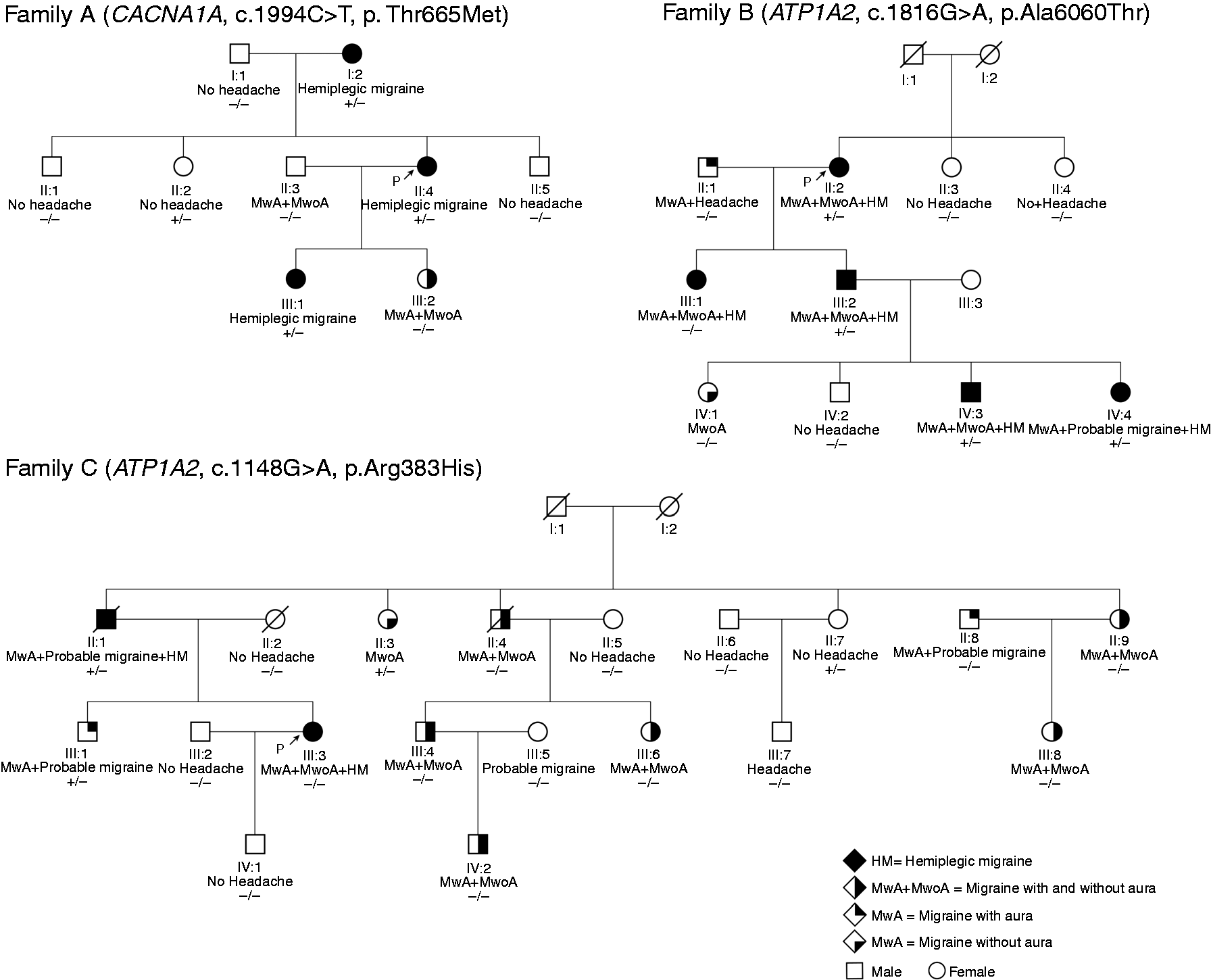
The other two likely causal mutations were located in the ATP1A2 gene. The p.Ala606Thr was present in one family co-segregating with the HM phenotype with a complete penetrance. Three of the mutation carriers have HM triggered by minor head trauma (Table 2). Interestingly, both WES and Sanger sequencing data confirmed that the proband's daughter does not carry the mutation, even though she has HM based on the ICHD-3 beta criteria (Figure 7, family B, III:1). However, her symptoms are less severe: for instance, she has motor weakness in the arm but not in the leg, she was 30 years old when she had her first migraine attack, and her attacks are not triggered by minor head trauma. Although her diagnosis could not be explained by the identified p.Ala606Thr variant, it could however be caused by other genetic factors combined with environmental factors. For instance, she might have inherited some still unknown genetic susceptibility factors from her father, who has migraine with typical aura.
The p.Arg383His variant in family C (Figure 7) was transmitted from an affected father to daughter and thus co-segregated with the HM phenotype. However, three unaffected family members (III:1, II:3, II:7) also carried the mutation. The proband's brother (III:1) has had one HM attack, and thus does not fulfill the IHS criteria for FHM (Table 2). Two other family members with the variant, but without HM, were over 40 years of age when they participated in the study (Table 2). Thus, it is unlikely that they would develop HM in later life. From those five family members with the p.Arg383His mutation, only two have an FHM diagnosis, corresponding to a 40% penetrance.
Furthermore, we identified one probably causal frameshift variant, p.Ser2501Ter, in the CACNA1A gene in two SHM individuals (Figure 8). The other patient has three relatives carrying the same variant and having diverse migraine symptoms, not fulfilling the HM criteria (Figure 8; Family E II:1, II:3, III:3). The variant is extremely rare in the gnomAD database (Table 1) and it is located in the last exon of the gene, where it creates a premature stop codon. Thus, in the protein level, the five last amino acids forming the end of the C-terminal region of the channel are missing.
Pedigrees of the Finnish hemiplegic migraine families with frameshift variant in the CACNA1A gene. Mutation allele is indicated with + and wild type allele with −; reference sequence: ENST00000360228.5 (CACNA1A).
Discussion
The present study describes one of the largest hemiplegic migraine samples screened for mutations in CACNA1A, ATP1A2 and SCN1A, and shows that HM patients do not seem to form a clearly separate group of their own, in relation to general migraine and aura symptoms, but rather reflect an extreme phenotype in the migraine with aura continuum. Only 7% (3/44) of the studied Finnish FHM families and none of the studied 201 SHM patients have likely damaging exonic mutations in the known three FHM genes.
Hemiplegic migraine is a clinically heterogeneous disease
HM patients reported more often severe and long lasting headache with extended aura episodes than NHMwA patients. They also had a higher proportion of attacks with visual aura symptoms and earlier age of headache onset compared to NHMwA patients. Thus, our clinical results are in line with the previously published results from Danish population-based studies (2,20).
In addition to other clinical characteristics, we found differences in the prevalences of self-reported psychiatric disorders, asthma, and Raynaud's phenomenon between the HM and NHMwA groups. There were no significant differences in prevalences of self-reported epilepsy and stroke between these groups. The prevalences seen in our study for epilepsy, diabetes and asthma are similar to prevalences reported for migraine with aura in a large population-based Danish study (21). However, the prevalence for stroke, which was not age-adjusted, was higher than in the Danish data (8.7% vs 1.7%).
Most HM patients had a positive family history for common forms of migraine. A notable proportion of SHM families (43%) had more than three family members with migraine with or without aura. The proportion was even higher in FHM families (78%). In some families, there were two SHM patients who were third- or greater-degree relatives. From this perspective, the distinction between SHM and FHM is ambiguous and might suggest that the genetic and overall pathogenic background is overlapping.
CACNA1A, ATP1A2 and SCN1A causative variants are present in only a small proportion of the hemiplegic migraine patients
Surprisingly, our results showed that only 7% (3/44) of the studied Finnish FHM families and none of the studied 201 SHM patients have exonic mutations in the known FHM genes. The percentage is lower, although of the same order of magnitude, than previously reported. The Danish population based studies showed that 14% of the studied FHM families (6/42) had mutations in the CACNA1A or ATP1A2 genes (6), and that these genes were not the major cause of SHM in a sample of 105 patients (7). Even though we did not identify any definitive FHM mutations in SHM patients, there are other studies supporting their role also in SHM (8). Patients with early-onset SHM associated with neurological symptoms are especially at high risk of carrying a causal mutation in CACNA1A or ATP1A2 (22).
One possible explanation for the lower percentage of mutation carriers compared to the Danish studies might be the selection process of patients. In the Finnish Migraine Genome Project, we have not specifically tried to recruit patients with HM but rather patients with all migraine subtypes. At the beginning of the project, we were focusing on migraine with aura families and, later on, families with all migraine subtypes. Another explanation could be our strict mutation criteria. In this study, we utilized large allele frequency databases and considered the identified variant as pathogenic only if the follow up analyses supported the conclusion.
Three identified pathogenic variants
All three pathogenic variants identified in this study have been described earlier, including supporting functional data on their damaging role. The p.Thr665Met is the most reported mutation in CACNA1A causing HM (3). The p.Ala606Thr is also a previously reported mutation (23) with functional data suggesting a loss of function effect (24). In our study, carriers of this mutation had a severe phenotype including FHM attacks triggered by minor head trauma and early age at onset of migraine (<14 year olds).
The penetrance of p.Thr665Met and p.Ala606Thr in our families is comparable to the previous reports on CACNA1A and ATP1A2 mutations (80–90%) (23,25). However, the penetrance of p.Arg383His is significantly lower, only 40% in this study, and the symptoms of the affected carriers are less severe. For instance, one of the carriers with HM reported experiencing his first migraine attack at the age of 27. He also reported mild headache intensity and 10–50 HM attacks at the age of 73 with short duration of hemiplegia (15 minutes). However, previous genetic and functional evidence (24,26), the very low population allele frequency and location in a highly conservative site all support the conclusion that p.Arg383His is also a true disease-causing variant.
In general, based on our observational results, clinical features of the patients with the identified causative variants are not clearly different from the other studied HM patients. They have a similar positive family load of common migraine and their attacks are severe, but not particularly more severe than the rest of the HM group. Because none of the identified mutations alone explain the observed clinical heterogeneity, this suggests that there are other genetic and/or environmental susceptibility factors for migraine in these families.
One probably causal variant in CACNA1A
Moreover, we identified one probably causal, extremely rare, frameshift variant (p.Ser2501Ter) in the CACNA1A gene. The variant creates a premature stop codon deleting the last five amino acids. The location in the very end of the transcript might make the deletion less damaging, but alternatively it might also protect the transcript from nonsense-mediated mRNA decay. Furthermore, the C-terminus of CaV2.1 α1A subunit is known to have a role in the regulation of the channel activity by participating in a number of protein–protein interactions (27). Interestingly, the very end of the C-terminal region contains a conserved DDWC-COOH (D2503-C2506) binding motif for the neuronal adaptor protein Mint1 (28). Lack of functional studies or more extensive genetic data makes interpreting the pathogenicity of this variant challenging, and thus additional reported HM patients and functional evidence is needed.
Likely benign variants
Four SCN1A variants (p.Arg1928Gly, p.Thr1250Met, p.Thr1174Ser, p.Arg542Gln) found in our study, previously reported in epilepsy or FHM3 (29–32), did not fulfill our predetermined mutation criteria, and thus were not classified as causative variants. Our strict mutation criteria are supported by a recent study re-evaluating the pathogenicity of SCN1A variants in common epilepsies and concluding that they are likely benign (33).
The p.Ala453Thr variant in CACNA1A has been suggested to be a negative modifier of the migraine aura phenotype, protecting its carriers from FHM in a migraine with aura family (34). Since the variant was identified in 12 of our families with HM and did not completely fulfill our predetermined mutation criteria, it was not considered here either as protective or causative.
Strengths and limitations
The main strength of this study is the large and well-characterized migraine family sample. Our study is part of the Finnish Migraine Genome Project, now consisting of over 2400 families and 10,500 individuals, which is, to our knowledge, the largest migraine family collection in the world. Even though our sample collection is not population based, the participants come from all geographical regions of Finland.
Because of the large number of participants, migraine phenotype data has been collected with a combination of individual interviews and with the extensive, validated Finnish Migraine Specific Questionnaire for Family Studies (9). The questionnaire is validated to diagnose migraine with aura and migraine without aura, but not specifically for hemiplegic migraine. To overcome this limitation, the participants were asked to give a precise description of hemiplegia during the attack, and in case of uncertainty the neurologist confirmed the diagnosis by phone.
Although questionnaire-based studies provide a number of advantages, it is also important to recognize that the self-reported data has its own limitations and is not equivalent to data derived from face-to-face interviews by headache experts. However, since the clinical information has been collected using identical methods regardless of the diagnostic subgroup, the same limitations apply to all patients, making the data comparable.
A low or unbalanced response rate causes non-response bias, thus decreasing the reliability of the specific comparison. In the present study, most of the questions had a high response rate (>80%), but there were also some questions with response rates lower than 60%. When comparing subgroups, the response rate in HM was somewhat higher than in the NHMwA group. The lower response rates for specific questions can be explained by our lengthy, extensive, and thematically structured paper questionnaire. If the respondent does not have the specific symptom or is uncertain about it, the question might be left unanswered. These factors might explain the lower response rate in the NHMwA group compared to the HM group. Our overall interpretation is that, when considering both the answers and the response rates, migraine has a greater impact on patients with HM compared to the patients with NHMwA. While the non-response bias has to be acknowledged, it should not influence the validity of the presented conclusions in a study of this scale and scope.
Due to the unique population structure of Finland, there is significantly less genetic variation compared to more mixed populations, especially in the rare and low allele frequencies. On the other hand, the rare and low frequency variants that are present are often enriched in the population (35). Since the Finnish population is a powerful resource for identifying disease-associated genetic variants, we expected to find new mutations in the FHM genes that would be specific to the Finnish population. However, this was not the case, since all the mutations identified in this study have been reported earlier in other European populations.
Future prospects
The fact that only 7% of the Finnish FHM families have likely causal mutations in the known FHM genes supports the hypothesis that there must be additional variants contributing to the HM phenotype. It is possible that causal mutations may locate in the noncoding regions of the known FHM genes, but it also suggests that there are additional genes contributing to the HM phenotype. Sequencing techniques might reveal rare and large effect variants in some new migraine genes, especially in families with several affected individuals.
An alternative hypothesis is that HM might not be a true monogenic disease, but a polygenic disease where common variants (36) contribute to the disease outcome. The familial load of migraine with and without aura, and the observation that HM patients do not cluster as a separate group, but rather have similar aura and headache characteristics to the patients with less severe phenotypes, suggest that there might be a shared genetic basis for all migraine subtypes. We speculate that, at least in some cases, this general genetic migraine load combined with additional genetic and environmental risk factors could cause the more severe attacks that the HM patients experience.
Supplemental Material
Supplementary figure 1 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary figure 1 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Supplemental Material
Supplementary figure 2 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary figure 2 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Supplemental Material
Supplementary figure 3 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary figure 3 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Supplemental Material
Supplementary table 1 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary table 1 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Supplemental Material
Supplementary table 2 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary table 2 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Supplemental Material
Supplementary table 3 -Supplemental material for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families
Supplemental material, Supplementary table 3 for The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families by Marjo Eveliina Hiekkala, Pietari Vuola, Ville Artto, Paavo Häppölä, Elisa Häppölä, Salli Vepsäläinen, Ester Cuenca-León, Dennis Lal, Padhraig Gormley, Eija Hämäläinen, Matti Ilmavirta, Markku Nissilä, Erkki Säkö, Marja-Liisa Sumelahti, Hanna Harno, Hannele Havanka, Petra Keski-Säntti, Markus Färkkilä, Aarno Palotie, Maija Wessman, Mari Anneli Kaunisto and Mikko Kallela in Cephalalgia
Footnotes
Article highlights
The results of the study highlight HM as a clinically and genetically heterogeneous disease.
Based on patterns of clinical presentation, HM patients do not seem to form a clearly separate group of their own, in relation to general migraine and aura symptoms, but rather reflect an extreme phenotype in the migraine with aura continuum.
Our study showed that only 7% of the Finnish familial hemiplegic migraine families have exonic mutations in CACNA1A, ATP1A2 and SCN1A genes, suggesting that there are additional genetic factors contributing to the HM phenotype.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Wellcome Trust [grant numbers WT089062, 098051 to A.P.]; the Academy of Finland [grant numbers 200923, 251704, 286500 to A.P., and 139795 to M.W.]; the Academy of Finland Center of Excellence for Complex Disease Genetics [grant numbers 213506, 129680]; the EuroHead project (LSM-CT-2004-504837); FP7-EUROHEADPAIN-no.602633; ENGAGE Consortium (grant agreement HEALTH-F4-2007-201413); EU/SYNSYS-Synaptic Systems [grant number 242167 to A.P.]; the Sigrid Juselius Foundation, Finland (to A.P.); the Folkhälsan Research Foundation, Finland (to M.W.); Medicinska Understödsföreningen Liv & Hälsa (to M.W.); the Helsinki University Central Hospital (to M.K., V.A.), and Beatriu de Pinòs (BP-A DGR 2010 grant) AGAUR, Generalitat de Catalunya (to EC-L).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.