
Abstract
Premise
The brain and the sensory nervous system contain a rich supply of calcitonin gene-related peptide (CGRP) and CGRP receptor components. Clinical studies have demonstrated a correlation between CGRP release and acute migraine headache that led to the development of CGRP-specific drugs that either abort acute attacks of migraine (gepants) or are effective as prophylaxis (antibodies). However, there is still much discussion concerning the site of action of these drugs.
Problem
Here we describe the most recent data related to CGRP in the trigeminal ganglion and its connections to the CNS, putative key regions involved in migraine pathophysiology. Gepants are small molecules that have limited ability to cross the blood-brain barrier (BBB), whereas CGRP antibodies are 1500 times larger molecules, and are virtually excluded from the brain, with a BBB permeability of < 0.1%. Thus we propose that the primary site of action for the antimigraine drugs is outside the CNS in areas not limited by the BBB.
Potential solution
Therefore, it is reasonable to discuss the localization of CGRP and its receptor components in relation to the BBB. The trigeminovascular system, located outside the BBB, has a key role in migraine symptomatology, and it is likely targeted by the novel CGRP drugs that successfully terminate migraine headache.
Introduction
Currently, the general view of migraine is that it is a frequent episodic disorder in which the central nervous system (CNS) has a key role, while the trigeminovascular system is necessary for the complete expression of peripheral symptoms and aspects of the headache (1). The CNS aspects of migraine were first shown by Weiller and colleagues (2), and later supported and extended by numerous imaging studies over the subsequent two decades (3). Recently, Schulte and May (4) revisited this idea of a central migraine generator using MRI (magnetic resonance imaging), by performing daily brain scanning of a subject over 30 days of the migraine cycle. They proposed that the origin of the migraine attack was in the hypothalamus-thalamus region. The authors observed that the response to trigeminal nociceptive stimulation varied over the cycle of the month and, in particular, that it increased in sensitivity 24 hours before the migraine attack. Brain activation during the early premonitory phase has also been observed when infusion of glycerol trinitrate was used to induce migraine-like attacks (5).
An alternative hypothesis for the generation of migraine attacks is the “vascular hypothesis”, which was based on early clinical observations that patients developed allodynia in the trigeminal projection areas and that different trigger factors, including some vasoactive agents given systemically, caused or exacerbated the symptoms and attacks (1). It is a challenging task for scientists to comprehend and connect these seemingly different views of the migraine attack. Often in science, however, there is an explanation that can incorporate several findings to an overall inclusive hypothesis.
CGRP release in primary headache conditions
Over the years there has been an intense search for biomarkers in primary headaches, starting with 5-hydroxytyptamine (5-HT) and its metabolites (6), amines, and much later neuropeptides (7). It was not until 1990 that a significant role of calcitonin gene-related peptide (CGRP) in migraine was first demonstrated by revealing that CGRP levels were increased in the cranial venous outflow during acute genuine migraine attacks (8). This result has subsequently been confirmed by several studies from different research groups. A striking finding was that CGRP levels normalized and the pain disappeared after subcutaneous sumatriptan both in acute migraine (9) and in cluster headache (10) patients. Interestingly, it was reported that systemic infusion of CGRP can cause migraine-like symptoms in migraine patients but not in otherwise healthy subjects (11). To understand this result, one must consider that (i) CGRP does not pass the blood-brain barrier (BBB) (12), (ii) the trigeminal ganglion has CGRP receptors (13) and (iii) the ganglion lacks a BBB, as does the dura mater (14,15), and it is accessible to systemic drugs (16). Hence, it is possible that systemic CGRP can enhance the activity within the trigeminovascular system and thus exacerbate migraine symptoms. In addition, the CGRP hypothesis is strongly supported by findings of the antimigraine effect of intravenous olcegepant (CGRP receptor blocker) and several other gepants (17).
For some time, it was hoped that CGRP could be used as a biomarker for migraine; however, this peptide is rapidly broken down in the blood (half-life < 10 minutes). Early migraine studies showed that significant release of CGRP into the external jugular vein (EJV) was not associated with an increase in the plasma level of CGRP in the cubital fossa vein (8,18). The issue of peptide analysis has been discussed to a great extent ever since. Technical issues, such as breakdown of CGRP and other peptides in the circulation, occurrence of peptide fragments, and antibody specificity, are important to keep in mind when presenting positive as well as negative results using different assays (19).
Defining the role of CGRP
For almost a century, the autonomic and sensory nerve supply to the intracranial vasculature have been of interest with regard to regulation of cerebral circulation and possible involvement in the pathology of stroke and migraine. However, progress in understanding the role of the perivascular innervation in the cranial circulation was initially hampered by the lack of suitable methodologies. The first useful methodology was the development of the histofluorescence method, which revealed the localization of amines and 5-hydroxytryptamine (5-HT) in nerve fibers. The work on defining the role of perivascular signaling molecules in cranial vasculature started with studies on noradrenaline and acetylcholine in sympathetic and parasympathetic nerves, respectively (20). The subsequent development of neuropeptide antibodies resulted in the first demonstration of the neuropeptide vasoactive intestinal polypeptide (VIP) in brain vessels (21). Subsequent work over the next 10 years demonstrated that a large number of neuropeptides were involved in cerebrovascular innervation (7). In particular, the finding that alternate expression of the human calcitonin gene could give rise to CGRP (22,23) led to the demonstration of CGRP in perivascular nerves in the brain (24) and in most other parts of the circulation (25,26).
Immunohistochemistry and quantitative radioimmunoassay methods were other methodological advancements that, in combination with specific denervation experiments, revealed the trigeminal origin of perivascular CGRP-containing nerve fibers (24). In addition, the trigeminal ganglion was described as containing small-mid sized neurons that store CGRP, while larger neurons contain CGRP receptors (Figure 1) (13). Detailed neuronal tracing in rat using a specific neuronal tracer (True Blue) placed in temporal, middle meningeal and cerebral arteries demonstrated movement of the tracer to a subpopulation of trigeminal neurons that contained CGRP (27–29). Interestingly, there was not a strictly unilateral distribution; some labelled fibers also reached the contralateral trigeminal ganglion. This is not unique to sensory fibers, but is also observed in the cranial sympathetic and parasympathetic systems (27–29). Subsequently, the use of specific tracers for sensory C-fibers (wheat germ agglutinin-conjugated horseradish peroxidase, WGA-HRP) and A-fibers (cholera toxin subunit b, CTb) revealed differential somatotopic localization in brainstem projection areas, the trigeminal nucleus caudalis, and the spinal C1-2 levels. We could differentiate the thin nociceptive (unmyelinated C - fibers and small myelinated Aδ – fibers, respectively) from the thick low-threshold mechanoreceptive myelinated Aβ- fibers (30–32). These fibers connected the middle meningeal (30), superior sagittal (31), temporal (32) and cerebral arteries (33) to specific brainstem regions. These tracer studies revealed the fiber connections, but not the neuronal messenger molecules involved. Research for > 30 years carefully mapped out several messenger molecules in the trigeminovascular system (7) and subsequent functional studies revealed the central role of the trigeminal sensory system in migraine. Among the candidate neuropeptides, CGRP has stood the test of time as a major transmitter molecule involved in primary headache disorders (7). The role of the other trigeminal messengers in headache, however, remains an enigma.
Distribution of CGRP, RAMP1 and CLR in trigeminal ganglion (TG) and sphenopalatine ganglion (SPG). The images show CGRP, RAMP1 and CLR single stainings, using FITC secondary antibodies (green colour) and blue DAPI nuclear staining.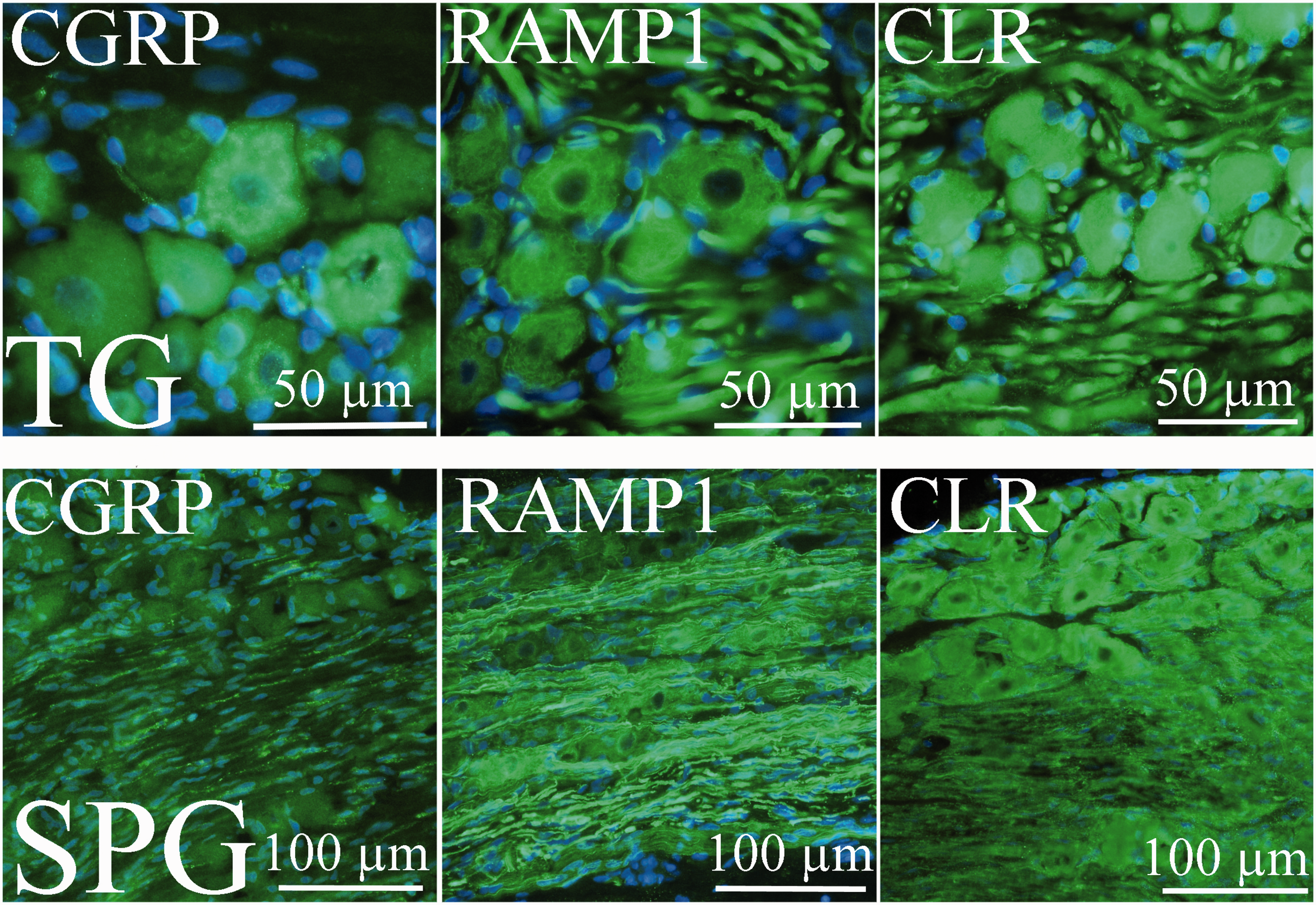
Early studies showed CGRP to be an astonishingly potent vasodilator that acts by stimulating vascular smooth muscle adenylyl cyclase (34). Thus, the response to CGRP is independent of a functional endothelium, a finding verified in most vascular regions examined to date (26). Activation of the trigeminal system results in antidromic release of CGRP (35) which acts on vascular CGRP receptors. The vascular responses have provided a useful method to characterize CGRP receptors and potential receptor blockers (36). The CGRP receptor is a G protein-coupled receptor of the B-type consisting of a calcitonin receptor-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) (37). The G-protein is coupled to the “receptor component protein (RCP) - adenylate cyclase” (38) to cause cAMP mediated vasodilatation in cerebral and meningeal arteries (39). Recently, detailed pharmacological and receptor binding studies revealed that the pattern of CLR/RAMP1 distribution and the mechanism of the gepant-type of CGRP antagonists are similar in resistance arteries from different species (40). There are also CGRP receptors on neurons and other cell types within the CNS and in the trigeminal ganglion, but the role of CGRP at these sites is poorly known. In the trigeminal nucleus caudalis/C1-2 levels of the brain stem, C-fibers storing CGRP, with or without co-transmitters, and A-fibers exhibiting CGRP receptor elements may function to transmit nociceptive signals to neurons that project centrally (41,42). Via the spinothalamic tract, ganglionic neurons project to regions in the brainstem and midbrain that then relay information to the thalamus and higher cortical regions (43).
We now know that CGRP is densely expressed in the central and peripheral nervous systems (Figure 2(a) and (b)). Two forms of CGRP are expressed in the body: αCGRP, which is prominently localized in neurons such as the primary spinal afferent C- fibers of sensory ganglia, and ßCGRP, which is the main isoform found in the enteric nervous system. In the CNS, αCGRP is widely expressed in cortical neurons, cerebellar Purkinje cells and hippocampal pyramidal cells, which is consistent with a role of CGRP in the modulation of nociception, motor function, secretion, and olfaction (26).
Overview of CGRP expression in the trigeminal vascular system (a) and in the CNS (b). Figure 2(a) shows fibers and cell bodies (in red) that express CGRP in the trigeminal ganglion and in the peripheral and central connections. The illustration (2(b)) shows CGRP expression in the CNS. There is a rich CGRP expression generally in grey matter and in the neuron, but not in fibre structures such as that seen in e.g. corpus callosum. Some of the CGRP-containing areas are shown in the image. Other transmitter circuits, 5-HT (serotonin), NA (noradrenalin), Ach (acetylcholine), dopamine and GABA (gamma aminobutyric acid), are also included in the image, visualizing the complexity of transmitter interactions.
Localization of CGRP receptors
Initial studies of CGRP receptor distribution with radiolabelled CGRP demonstrated that CGRP binding sites were widely expressed throughout the rodent brain (44). The discovery of the CGRP receptor elements, CLR and RAMP1 (37), advanced the field and allowed creation of specific antibodies towards these components. Use of the antibodies in immunohistochemical studies revealed a more detailed distribution of the CGRP receptor in migraine-related and other structures in the brain (Figure 3(a) and (b)). Another study on human cerebral and middle meningeal arteries demonstrated the presence of CLR and RAMP1, 2 and 3 in the vascular smooth muscle cells (45). When all three RAMP isoforms are expressed, it appears that RAMP1 determines the receptor phenotype, consistent with a functioning CGRP receptor. Recent work has mapped in detail the distribution of CLR and RAMP1 to the cytoplasm of trigeminal neurons, at peripheral sites on the intracranial vasculature (in the smooth muscle cells), in the dura mater (both vascular and avascular localization) and in the brainstem (13,36,41,46). In the trigeminal ganglion, most CGRP-containing neurons (small to medium sized) do not contain the receptor elements; instead CLR and RAMP1 are co-expressed on CGRP-negative, large neurons and on satellite glial cells, which suggests the possibility of intra-ganglionic communication (47). Consistent with the early functional findings reported in 1985 (34), the human study (36) did not find CGRP receptor components in endothelial cells. This also correlates with functional studies in human intracranial arteries where removal of the endothelium or blocking endothelial factors (NO, PGI2 or EDHF) did not modify CGRP-induced relaxation (34). Detailed mapping of the location of CGRP and its receptor in peripheral and central sites has now been done in several species, including primates such as monkey and human (13,46,48). The expression of CGRP and the CGRP receptor is astonishingly complex and suggestive of many different brain functions.
Overview of CLR and RAMP1 in the trigeminal vascular system (a) and in the CNS (right). (a) In the trigeminal vascular system CLR expression is shown in pink and RAMP1 expression in blue. (b) The pattern resembles that of CGRP expression, but CGRP does not colocalize with the receptor elements. In the CNS, RAMP1 and CLR are found in all fibre structures (green). In grey matter and nuclei, the receptor components are observed in fibers intermingled between the neurons (lined green).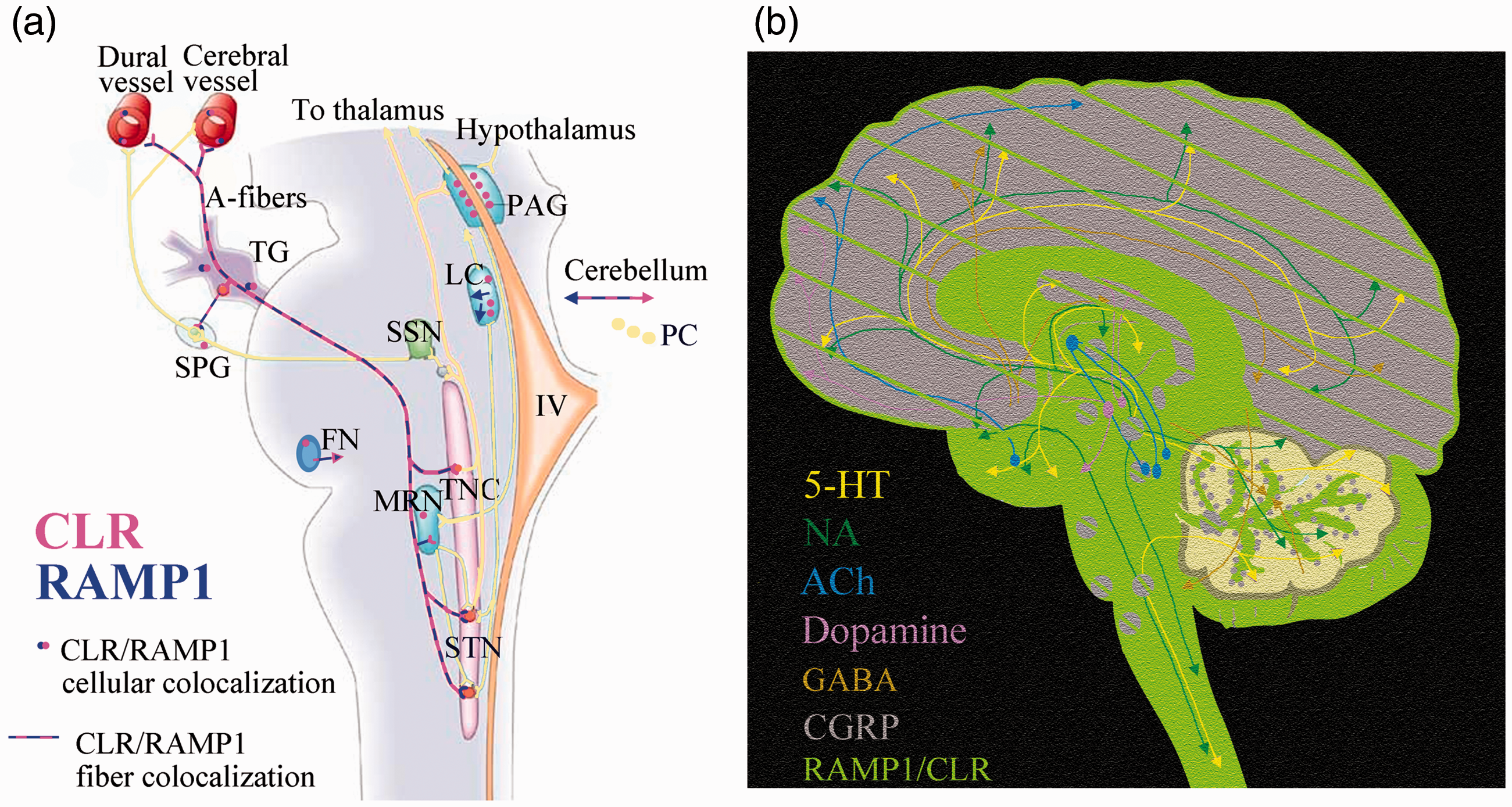
CGRP in the brainstem
Clinical studies using imaging techniques such as PET and functional MRI (fMRI) have highlighted several brain regions that are activated in different phases of the migraine attack (2,4,5). Particular interest has been directed at the midbrain, pons, substantia nigra, red nucleus, periaqueductal gray (PAG), nucleus raphe magnus (NRM) and the locus coeruleus (LC). Based on these findings we examined the CNS distribution of various components of the CGRP system (48). There was rich expression of CGRP in striatum, amygdalae, colliculi and cerebellum, as well as in the cortex (48,49).
The use of novel specific antibodies towards CLR and RAMP1 in the primate brain further reinforced our realization that the role of CGRP and CGRP receptors in the CNS is very complex (48). In the primate, CLR and RAMP1 mRNA and protein expression are found in the pineal gland, the medial mammillary nucleus, periaqueductal grey (PAG), area postrema, the raphe nuclei, the gracile nucleus and spinal trigeminal nucleus (50). This study thus reveals numerous potential CNS CGRP targets. Moreover, the CGRP receptor components do not always occur together, which can be indicative of receptor subtypes that bind other molecules of the CGRP family such as calcitonin, amylin and adrenomedullin.
Within the cerebellum, Purkinje cell bodies store large amounts of vesicular CGRP, and the CLR/RAMP1 complex is expressed both in the cell body and in axons and dendrites (51,52). The cerebellum is important for modulating many cortical motor and sensory inputs (53). It exerts an inhibitory control over the cerebral cortex, and thus may play an important role in filtering sensory inputs (51). Early studies of acute migraine attacks by PET revealed activation of cerebellar regions (2,54). However, these studies did not provide any deeper explanation for the activation that was observed.
The general view today suggests that a migraine attack starts in the CNS (2,55) and spreads to the trigeminovascular system in order to evoke all the facets of a migraine attack. Because the triptans and gepants can modify the acute attacks, it is logical to hypothesize that there are sites within the CNS that can be modified by these drugs. Using a brain penetrant PET tracer, binding to CGRP receptors was most obvious in the cerebellum (56). However, the CGRP blocker telcagepant did not modify the PET tracer binding when administered in a clinical dose that successfully aborts a migraine attack. A 10 times higher dose was necessary to compete away the tracer binding, but this dose had no better efficacy in migraine. Thus, the inability of clinical active drug concentrations to displace central CGRP bindings sites points to the alternative hypothesis that the antimigraine site of action of telcagepant lies outside the CNS.
The trigeminal ganglion has no BBB
Antibodies towards CGRP and the CGRP receptor have provided new tools that may assist in explaining how anti-migraine drugs work (16). These molecules are relatively large, and therefore have little chance to pass the BBB in effective doses. In fact, available data suggest that the percentage of triptans and gepants that pass the BBB is 3% and 2%, respectively (16). Antibodies are about 1500 times larger in size, which suggests that their ability to pass the BBB is much less, around 0.1 – 0.01%. Gepants and antibodies to CGRP and CGRP receptors are all therapeutically effective in migraine, which suggests that they share a common site of action, likely located outside the BBB.
For decades, it has been known that the dura mater and the middle meningeal artery are devoid of a BBB, however this was not generally known for the trigeminal ganglion. To study whether the trigeminal ganglion could be a possible site of action for CGRP receptor antagonists, we performed a series of experiments to test for a BBB in this tissue. We used Evans blue dye, which binds to plasma albumin in the circulation to form a large complex that is excluded from the CNS by the BBB. The dye was found to be richly distributed in the trigeminal ganglion (14). Thus, the trigeminal ganglion, like the dura mater (15), lacks a BBB and is freely accessible to circulating compounds. This conclusion was further supported by a recent study using another tracer, labelled EDTA, which allows for quantitative calculation of transfer constant. The trigeminal ganglion was found to be > 100 times more accessible to the passage of this tracer than the cerebral cortex, cerebellum, brainstem, and trigeminal nucleus caudalis (15). Therefore, we propose that current antimigraine drugs may reach sites in the dura mater and the trigeminal ganglion, but not in the CNS apart from regions lacking a BBB. Moreover, there is no clinical evidence for alterations in the BBB during migraine attacks (57). Goadsby and colleagues recently addressed this issue and provided information in support of our view (58). Thus, it is likely that the main response to antimigraine drugs occurs in extracerebral sites and in regions not protected by the BBB.
Conclusion
CGRP-antagonism is an effective treatment mechanism in migraine. There is evidence that the newly developed anti-CGRP-drugs act peripherally (i.e. outside the BBB) and not centrally. This seemingly contradicts the hypothesis of migraine being of central origin and that a lot of known migraine prophylactics probably act centrally. This needs to be discussed – how can a peripheral blockade change the frequency of a centrally-driven disease? Central mechanisms might lead to trigemino-nociceptive disinhibition and thus to a broader CGRP action in areas outside the BBB, which contributes to the development and sustainment of migraine pain. The main site of action of CGRP/its receptor antibodies might be the trigeminal ganglion.
Article highlights
Defining the role of CGRP Localization of CGRP receptor components CGRP in the brainstem The trigeminal ganglion has no BBB
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by grants from the Swedish Research Council (no 5958) and the Swedish Heart and Lung foundation.