
Abstract
Background
Calcitonin gene-related peptide (CGRP) is a key molecule in migraine pathophysiology. Most studies have focused on CGRP in relation to migraine without aura (MO). About one-third of migraine patients have attacks with aura (MA), and this is a systematic review of the current literature on CGRP and MA.
Methods
We performed a systematic literature search on MEDLINE for reports of CGRP and MA, covering basic science, animal and human studies as well as randomized clinical trials.
Results
The literature search identified 594 citations, of which 38 contained relevant, original data. Plasma levels of CGRP in MA patients are comparable to MO, but CGRP levels varied among studies. A number of animal studies, including knock-ins of familial hemiplegic migraine (FHM) genes, have examined the relationship between CGRP and cortical spreading depression. In patients, CGRP does not trigger migraine in FHM, but is a robust trigger of migraine-like headache both in MA and MO patients. The treatment effect of CGRP antagonists are well proven in the treatment of migraine, but no studies have studied the effect specifically in MA patients.
Conclusion
This systematic review indicates that the role of CGRP in MA is less studied than in MO. Further studies of the importance of CGRP for auras and migraine are needed.
Introduction
Migraine is a neurovascular headache (1) thought to arise from a primary brain dysfunction, leading to activation and sensitization in the trigeminovascular system (2) and the release of vasoactive neuropeptides such as calcitonin gene-related peptide (CGRP) (3).
CGRP is a 37-amino-acid neuropeptide identified in the early 1980s (4,5) that belongs to a structurally conserved family of peptides that includes calcitonin, adrenomedullin and amylin (6).
CGRP is broadly distributed in the nervous system (6,7) and in the trigeminal pain pathway at peripheral (8–10) and central levels (11–13). Rosenfeld and colleagues (5) reported the presence of CGRP immunoreactivity in the rat trigeminal ganglia and Mason et al. (14) demonstrated calcium-dependent CGRP release upon depolarization from trigeminal ganglia in vitro (15).
The blood vessels of the meninges (dura and pia) are richly innervated by CGRP-containing fibers originating in the trigeminal ganglion (9,16,17), and CGRP is a potent vasodilator (18) both in cerebral and extracerebral arteries (16) (see also Figure 1).
The trigeminovascular system, with focus on calcitonin gene-related peptide (CGRP) signaling. Left: CGRP released from perivascular afferents in the dura, causes dilation of arterial vessels (AV). Nitric oxide (NO) from the vascular endothelium facilitates CGRP release. Middle: In the trigeminal ganglion, CGRP signals to trigeminal ganglion neurons (Aδ/C) expressing CGRP receptors. Right: In the trigeminal nucleus, CGRP facilitates nociceptive transmission to second-order neurons possibly by increasing the release of the excitatory neurotransmitter glutamate (Glu) from neighboring primary afferent terminals. CGRP may also signal directly to second-order neurons (dotted arrows). Figure adapted from Messlinger et al. (19) and used with permission.
CGRP is released from perivascular trigeminal nerve terminals (20,21), but does not activate or sensitize the nociceptors in the rat meninges (22) or in human skin and muscle (23). Animal models of headache and pain suggest modulatory role of CGRP in nociceptive transmission (22,24–27).
Studies in migraine patients showed elevation of CGRP during (28) and outside of migraine attacks (29). However, Tvedskov et al. found no changes in plasma CGRP during migraine attacks compared to outside of attacks in patients with migraine without aura (MO) (30) and thus challenged earlier reports.
Human studies supported the importance of CGRP in migraine pathogenesis. With the use of an experimental migraine model (31), Lassen et al. (32) showed in a double-blind crossover study that CGRP-infusion induces a migraine-like headache in 50% of MO patients. Thus, evidence from basic science and animal and human studies pointed to CGRP antagonism as a potential target for antimigraine drugs. Large randomized controlled trials have now indeed confirmed that CGRP antagonists are effective in treating acute migraine attacks. The relationship between CGRP and migraine is therefore a matter of intense research, and has been extensively reviewed (see e.g. Raddant and Russo (33), Ho et al. (34), and Villalón and Olesen (35)). The exact pathways involved in CGRP-induced migraine attacks and mechanisms of action of CGRP antagonists are not fully clarified but may involve both peripheral and central site of action (13,36).
Most studies have focused on CGRP in relation to MO, and CGRP seems to be primarily involved in the headache component of migraine. About one-third of migraine patients have attacks with aura (MA) (37); transient neurological symptoms attributed to brain dysfunction (38), likely to be the symptom of cortical spreading depression (CSD) (39) originally described by Leão (40). Although MA and MO may display a similar headache phase (41), the head pain after aura attacks seems to be milder and shorter lasting compared to MO (42). A possible difference in CGRP mechanisms between the two major types of migraine has largely been ignored.
We performed a systematic review of the current literature on CGRP and MA. Our aim was to find all relevant scientific data on CGRP and MA, including clinical subtypes of MA.
Methods
Literature search
We performed a systematic literature search identifying articles reporting original data on CGRP and MA or CSD in patients with clear diagnoses of MA, including subtypes, according to the guidelines of the International Headache Society (IHS) (41). The literature search was concluded September 1, 2013, and was based on PubMed using the keywords “calcitonin-gene related peptide” and “CGRP” in combinations with “migraine,” “migraine aura,” “migraine with aura” and “cortical spreading depression.” All papers with an abstract in English were included, including basic science and animal and human studies. References of review papers, editorials and other references without original data were scrutinized. We also considered articles from the reference list of studies that were found to be relevant as well as literature that was known to be relevant by the authors.
Data extraction
Both authors examined the abstracts found in the literature search. Whenever the title or abstract suggested that relevant data could be part of the publication, the entire manuscript was read. Papers with unclear diagnoses of MA after the current IHS criteria (41) were not included, and papers describing mixed migraine diagnoses were included only if a substantial number of patients had MA.
Results
The flowchart for selection of papers included in this review is shown in Figure 2, following Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines. The search strategy identified 594 papers. We excluded 259 papers that were either review papers, editorials or other references without original data and 297 papers that did not contain relevant data on CGRP, MA, CSD or without clear IHS diagnoses or unclear methodology (data search and eligibility score sheet on file). We included 38 papers for the review (28,29,43–78).
Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) flowchart of retrieved, excluded and analyzed papers with data on calcitonin gene-related peptide (CGRP) and migraine patients with aura (MA).
MA and plasma levels of CGRP
Levels of circulating CGRP in MA patients. If studies included MO patients or control subjects, these values are presented for comparison.

CGRP: calcitonin gene-related peptide; MA: migraine with aura; MO: migraine without aura; Migraine: MA and MO; ND: no data; NS: not significant.
Outside of attack
Seven studies reported interictal CGRP levels in adult MA patients (29,45,58,63,68,76,77). Four studies reported that CGRP levels are increased in migraineurs compared to control subjects; three studies found no difference.
Two studies reported similar CGRP levels in MO and MA patients (45,58). In contrast, the newest study based on a fairly large female cohort (77) reported that in patients with chronic migraine, patients with aura had higher CGRP levels than patients having only MO, whereas no difference was found for episodic migraine. Both groups had higher CGRP levels than controls. Interestingly, one study reported that CGRP levels in the carotid artery and internal jugular vein were elevated compared to reference peripheral blood levels in MA patients. The same authors found no increases of CGRP in internal jugular venous blood during the aura phase of migraine attacks and no difference between cerebral venous and arterial CGRP levels, speaking against a net release of CGRP from the brain (70). One study reporting increased interictal CGRP levels in migraineurs (n = 20) found more pronounced CGRP elevations in MO patients (n = 14) than in patients who also reported occasional MA attacks (n = 6) (29).
During attack
Four references were found (28,65,68,70) (see Table 1). Two studies compared ictal to interictal CGRP levels in the same patients and found either unchanged (70) or increased (68) CGRP levels. Unchanged CGRP levels in internal jugular venous blood were reported during the aura phase in MA patients (n = 8) referred for cerebral angiography, and the procedure provoked attacks of MA in four patients (70). CGRP concentration levels were elevated above control values from the literature, but did not differ among patients experiencing aura or not and no difference between cerebral venous and arterial CGRP levels, speaks against a net release of CGRP from the brain (70).
CGRP was increased both in MA and MO patients, with significantly higher CGRP levels within two hours of onset of symptoms compared to three and four hours (68), suggestive of CGRP increase early in the attacks. This study also reported increased CGRP levels in the peripheral circulation during migraine compared to headache-free intervals. This finding contrasted with that of Goadsby et al., who found no increase of CGRP in cubital venous blood but increased jugular venous levels during a migraine attack (28). This study did not investigate the interictal CGRP levels in the same patients.
Two recent studies found that CGRP levels increased during migraine attacks compared to the interictal state but data were either not reported separately for MO (n = 11) and MA (n = 8) (44), or were based mainly on MO patients (79).
Antimigraine treatment effect on CGRP levels
CGRP is released from trigeminal neurons expressing the target of triptans, the 5-HT1B/1D receptors (80). One study compared CGRP levels during a migraine attack before treatment and two hours after rizatriptan administration in 33 patients (MO = 27, MA = 6).
CGRP levels decreased significantly both in MO and MA after treatment compared to attack levels, without differences between MA and MO (65). In a group of eight patients (72) (MO = 6, MA = 2) headache-phase CGRP level for the group was elevated 60 ± 8 pmol/l (reference < 40 pmol/l). In responders to treatment (n = 7, MA = 2) CGRP level dropped to within the normal range with a post-treatment mean of 40 ± 8 pmol/l for the entire cohort.
For this review we excluded a study that measured CGRP in saliva as a noninvasive method to assess CGRP as a biochemical marker of trigeminal nerve activation in patients with MO and MA, as the exact diagnoses were not disclosed (81). We excluded one paper examining CGRP levels in saliva before, during and after treatment with rizatriptan in a mixed migraine population, as data for MO and MA were not reported separately (82). Based on the methodology, we also excluded an animal study reporting that electro-acupuncture was able to suppress KCI-induced CSD and reduce plasma CGRP (83). A study of pediatric patients (84) found no difference between MA and MO, even though CGRP in migraineurs tended to be higher than in controls, but the study did not present separate data for attacks and interictal state, and was not included in the review. In a study of 40 patients (without migraine history) undergoing transcatheter atria-septal defect (ASD) closure; 24 patients reported new-onset migraine headache after closure, 13 of these (54%) reported visual aura. CGRP was increased during attacks, but data for MA patients were not reported separately (85).
CGRP and CSD
Transient arteriolar dilation and cerebral hyperemia associated with CSD is caused both by nitric oxide (NO) and CGRP (69,74,75) probably released from perivascular nerves. CSD did not, however, change CGRP levels in the jugular vein in cats (71), and CSD-like conditions did not alter the release CGRP from the dura in rats (67). CGRP was not increased in internal jugular venous blood during the aura phase of a migraine attack in four patients (70). Recent data suggest an important role for CGRP in CSD as it was reported that CGRP receptor antagonists could block CSD (44). CGRP application did not, however, induce CSD (44).
Genetic susceptibility, CGRP and MA
Genetic association studies are widely used for identifying susceptibility genes in complex disorders such as migraine. We found five studies (43,53,62,73,78) looking at the association between genetic variation, CGRP and MA.
An association study in 278 migraineurs (85% MA) of the relation between migraine and a well-described single-nucleotide polymorphism (SNP) in the CGRP gene showed no significant association (53).
An association study that included 284 migraineurs found no relation between migraine and SNPs in the promoter for the CGRP gene (228 patients, 64% MA) nor in the receptor activity-modifying protein 1 (RAMP1) of the CGRP receptor (two SNPs were examined in (243 patients, 60% MA and 226 patients, 61% MA, respectively) (43).
A case-control study of 188 migraine patients (41% MA) examining the relationship between CGRP brain-derived neurotrophic factor (a gene that is differentially expressed after CSD) and CGRP found no significant differences between cases and controls regarding the SNPs analyzed (73).
Adenosine A2A receptor (A2AR) modulates the effect of CGRP in the brain, and activation of the receptor leads to excitatory actions of CGRP on synaptic transmission in the hippocampus and N-methyl-D-aspartate (NMDA) receptor-mediated transmission, and thereby possibly CSD (86). A case-control study in 265 migraine patients (46% MA) found a six-marker haplotype A2AR gene variation was more frequent in MA but not in MO, compared with a matched control group (62). The association between the alpha CGRP gene polymorphism CALCA T-692C was examined in 134 female migraineurs and 96 healthy female controls. The genotype and allele frequencies of the CALCA T-692C gene polymorphism did not differ between the migraine and control groups nor between MA and MO patients (78).
Effect of systemic CGRP in patients with MA
A number of studies have examined the effect of CGRP administration in patients with MA.
MA
Fourteen patients suffering exclusively from migraine with typical aura (MA) according to the IHS criteria (41) were included in a controlled provocation experiment (56). The major outcome of the study was that systemic administration of CGRP (1.5 µg/min CGRP over 20 minutes) induces more migraine-like attacks in MA patients than controls; 57% of MA patients reported migraine-like attacks compared to none in controls, and one in four patients (28%) also reported aura symptoms (56).
Familial hemiplegic migraine (FHM)
Nine FHM patients with identified FHM mutations were recruited from a population-based cohort and included in a controlled provocation experiment.
CGRP (1.5 µg/min CGRP over 20 minutes) failed to induce more auras or more migraine-like headache in FHM patients than in healthy controls (60). Given the disparity of migraine-induction in FHM patients (60) and MO/MA patients (32,56), the migraine-inducing effect of CGRP was next examined in the large group of FHM patients in whom genetic mutations were ruled out. For this study, 11 FHM patients without known mutations were recruited along with 11 healthy volunteers. CGRP (1.5 µg/min CGRP over 20 minutes) failed to induce more migraine-like attacks in FHM patients than controls (49). No FHM patients reported aura symptoms after CGRP.
CGRP in animal studies of MA
An interesting addition to the armamentarium of animal models of MA is based on knock-in of genes from FHM: a dominantly inherited subtype of MA, phenotypically characterized by fully reversible half-sided weakness and other aura symptoms (41). FHM mutations seem to predispose to CSD (87) and interestingly, both CACNA1A (FHM-1) and ATP1A2 (FHM-2) knock-in mice have showed increased susceptibility to CSD (88,89).
Mathew et al. (48) investigated how the FHM-1 mutation affects CGRP expression in migraine-relevant central nervous system (CNS) structures, such as the trigeminocervical complex, the trigeminal ganglia and in dorsal root ganglia. The FHM mice had a significantly lower percentage of CGRP-immunoreactive neurons compared to wild type both in the trigeminal ganglion and the trigeminocervical complex. In the same mouse model, CGRP release from the trigeminal ganglion was larger in FHM-mutants than wild-type mice, but the mutation did not alter depolarization-evoked CGRP release from the dura mater (47). Based on this result, the authors suggested that in FHM-1, the facilitation of peripheral mechanisms of CGRP action, such as dural vasodilation and peripheral sensitization at the meninges, does not contribute to the generation of headache. These somewhat surprising results could be related to the reduced sensitivity of FHM patients to CGRP infusion (49,60).
Interestingly, in trigeminal cell cultures, FHM-mutant mice release more CGRP than the wild type, both under basal conditions and after stimulation with bradykinin, which also increased the percentage of glial-responding cells in KI culture compared to the wild type (51).
CGRP antagonists in treatment of MA
CGRP antagonists in randomized clinical trials with MA patients.
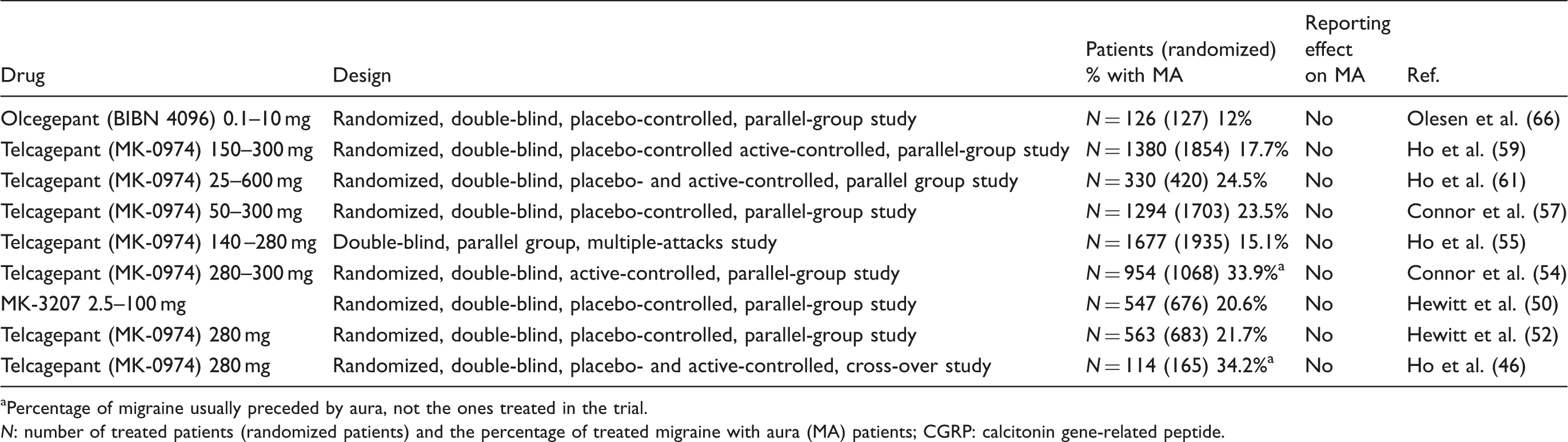
aPercentage of migraine usually preceded by aura, not the ones treated in the trial. N: number of treated patients (randomized patients) and the percentage of treated migraine with aura (MA) patients; CGRP: calcitonin gene-related peptide.
The total number of randomized patients was 8222, of whom 7394 were treated (90%). Based on the seven trials that reported treatment data with respect to aura, 19.3% (12% to 24.5%) had MA. None of these studies (46,50,49,52,54,55,57,59,61,66) reported possible treatment effects on aura frequency or migraine headache after aura.
In the study of the selective intravenously (IV)-administered CGRP antagonist olcegepant BIBN 4096, the percentage of treated attacks with aura was 12% (66), the number of patients with a history of aura attacks in the study was, however, somewhat higher (30.5%), similar to data from the two trials giving patient-reported data on aura based on patient migraine history (46,54).
For this review, we found no clinical trials that examined the effect of CGRP-antagonists exclusively in MA patients. We included only clinical trials for which clear distinction was made between MA and MO, as recommended by the IHS clinical trials subcommittee (90).
Three randomized trials of oral CGRP antagonists, BI 44370 TA (91), BMS-927711 (92) and telcagepant, MK-0974 (93), examined MO and MA patients, but were not included in the review as the publications did not report the number of patients with MA. We did not include a paper presenting the pharmacokinetic data of the olcegepant trial (94) and two papers presenting post-hoc analyses of the telcagepant trials (95,96).
One study examined the effect of intranasal civamide for the acute treatment of migraine headache (97). Civamide is a vanilloid receptor agonist and neuronal calcium channel blocker that inhibits the neuronal release of neurotransmitters including CGRP. The trial included 37 patients (16.7% MA), but results were not reported separately for MO and MA, and levels of CGRP were not analyzed.
Discussion
As expected, there is an extensive body of research on the anatomy, physiology, nociception and pharmacology of CGRP and the trigeminovascular system. We have included papers with a focus on CGRP and MA, CSD or CGRP-based treatment of MA patients.
MA and plasma levels of CGRP
Most studies report similar interictal plasma levels of CGRP in MA and MO (45,58,63,68,76,77), even though CGRP levels varied among studies. There is a need for a good, prospective well-powered study to clarify if CGRP is increased in MA during aura and ensuing migraine.
Antimigraine treatment effect on CGRP levels
The potential relation between headache, CGRP levels and treatment effect have been reported for paroxysmal hemicrania (98), cluster headache (99,100) and MO (101–103). These studies suggest that CGRP is elevated during attacks and successful treatment leads to normalization of CGRP. Data from MA patient are still too limited to draw firm conclusions, as we found only two papers with a total of eight MA patients (65,72).
CGRP and CSD
Recent studies suggest that CSD can lead to a long-lasting increase in activity of dural afferents and of trigeminovascular neurons in the trigeminal nucleus caudalis, indicating that CSD can activate the trigeminovascular system and potentially lead to headache (104–106). This effect, however, is unlikely to be mediated by CGRP because CSD does not lead to increased release of CGRP (67,71). Repetitive depolarizations in the brainstem, the region of the proposed migraine generator, do not spread to the cortex to cause CSD and do not affect CGRP release from nociceptive nerve endings in the dura mater (107). CGRP was not increased in internal jugular venous blood during the aura phase of migraine attack in four patients (70). One recent study challenges these data, as it was reported that CSD lead to a calcium-dependent release of endogenous CGRP in rat cortical slices after KCl application (44). The same study suggests that CSD inhibition can be achieved through treatments that block CGRP receptors. Whether this effect exists in humans has yet to be proven. More work is needed to clarify the possible link between CGRP, CSD and MA.
Effect of systemic CGRP in patients with MA: Provocation studies
The study by Lassen et al. (32) proved in a realistic human model that CGRP infusion caused migraine in MO patients and thus can be causative for migraine. It is therefore noteworthy that the first attempt to use CGRP to trigger migraine in MA patients failed: CGRP did not induce more migraine headache in FHM-patients than in healthy controls (49,60). In contrast, CGRP infusion was a robust trigger of migraine-like headache in MA patients (56), suggesting a reduced sensitivity in FHM to CGRP compared to MO and MA and possibly shared migraine mechanisms in MA and MO.
CGRP administration did not induce any aura symptoms in MO and FHM patients. In MA, however, one in four MA patients (28%) reported visual aura following CGRP infusion (56). How CGRP might induce visual aura is not clear since CGRP infusion has no direct effect on blood oxygenation level-dependent (BOLD) activity in the visual cortex in healthy controls (108).
Genetic susceptibility, CGRP and MA
Genetic studies have provided no convincing link between CGRP and MA. FHM has been widely regarded as an important genetic model to study the pathogenetic mechanisms both in FHM and common types of migraine (109,110).
The finding of reduced sensitivity to CGRP in FHM compared to MO and MA, combined with the phenotypic difference in FHM and typical MA, suggests that results from FHM may be less relevant to understanding typical MA than initially hoped. Indeed, most FHM studies seem to indicate different molecular pathways across the migraine spectrum and that understanding of molecular heterogeneity is important for future migraine research.
CGRP antagonists in treatment of migraine
Preclinical studies
The first CGRP receptor antagonists were based on peptide fragments of CGRP (111) and were used to examine vasodilation and neurogenic inflammation in animals (112), but turned out to have a low in vivo potency (113). Based partly on high throughput screening, a number of selective CGRP antagonists have been developed (114–118).
The exact site of action of CGRP and its antagonists is still highly debated. In healthy volunteers, olcegepant (BIBN 4096) effectively prevents CGRP-induced headache and extracerebral vasodilation without aborting CGRP-induced increase in cerebral blood flow (CBF) and a possible dilation of the middle cerebral artery (119).
These data suggest an extracerebral site of action of the antagonists, and contrasts with findings in animals that CGRP antagonists may act centrally (120), at least at the level of the trigeminocervical complex (121) to inhibit trigeminovascular activation (36).
In rats, IV olcegepant reduced neuronal activity in the spinal trigeminal nucleus, a likely marker of activity in the central trigeminal nociceptive pathway, supporting a central action of CGRP antagonists (122). A recent positron-emission tomography (PET) study in healthy volunteers reported a low central CGRP receptor occupancy after clinical doses of a CGRP antagonist (10).
Clinical studies
There is clearly a need for new treatment modalities in migraine, especially MA. CGRP antagonists have a highly specific antimigraine effect and no vascular effects that could limit their use in MA patients (123) or cardiovascular comorbidity (124,125). They do, however, have a relatively low efficacy (126) and the role of this highly interesting drug class is still under investigation.
The development of telcagepant and MK-3207 was halted in phase III after clinical trials induced a higher concentration of liver transaminases (127). Enhanced formulations of oral CGRP receptor antagonists are currently in development, and have shown promising treatment results in double-blind, randomized, placebo-controlled trials (50,91). The CGRP antagonists abort migraine pain and improve the associated symptoms of nausea, phonophobia and photophobia.
Whether CGRP antagonists can be used to reduce aura frequency has not yet been evaluated prospectively, but since CGRP receptor antagonists may block CSD, this seems possible (44) and should be studied in future trials. Whether CGRP antagonists have the same antimigraine efficacy for MA and MO attacks have not been published in any of the many randomized clinical trials, but could be a focus for post-hoc analyses of the current data as well as the focus for future trials.
Conclusion
This systematic review suggests that more work is needed to clarify the possible link between CGRP, spreading depression and migraine aura. Conflicting results regarding circulating CGRP levels in MA, and the lack of studies reporting the effect of antimigraine treatment on CGRP levels, suggest a need for new, prospective studies. Clinical trials have yet to examine the effects of CGRP antagonism on migraine aura, and the importance of migraine aura on treatment outcome.
Clinical implications
About one-third of migraine patients have attacks with aura (MA), and this is a systematic review of the current literature on calcitonin gene-related peptide (CGRP) and MA. Plasma levels of CGRP in MA patients are comparable to migraine without aura (MO), but CGRP levels varied among studies. In patients, CGRP does not trigger migraine in familial hemiplegic migraine, but is a robust trigger of migraine-like headache in MA patients. The treatment effect of CGRP antagonists are well proven in the treatment of migraine, but no studies have studied the effect specifically in MA patients. More work is needed to clarify the possible link between CGRP, spreading depression and migraine aura.
Footnotes
Funding
This work was supported by The Lundbeck Foundation for Neurovascular Signalling (LUCENS); the Research Foundation of the Capital Region of Denmark; the Danish Council for Independent Research-Medical Sciences (FSS; grants 271-08-0446 and 12-127798); and the Novo Nordisk Foundation.
Conflict of interest
None declared.