
Abstract
Background
The syndrome of transient headache and neurologic deficits with cerebrospinal fluid lymphocytosis (HaNDL) is a self-limited benign disorder of unclear pathogenesis, with diverse clinical manifestations.
Cases
We report two unusual presentations of this entity. The first case developed a catastrophic picture, characterized by acute elevation of intracranial pressure, necessitating emergency life support. The second case presented with hyperacute-onset mixed aphasia and facial droop, masquerading as acute ischemia of the middle cerebral artery territory. Both patients made full recoveries.
Conclusion
The possibility of HaNDL should be considered in individuals presenting with unusual patterns of headaches and transient neurological symptoms. Our report will expand the spectrum of this disorder, which will help physicians not only to recognize the unusual manifestations of this rare disorder, but also consider potential therapeutic interventions.
Keywords
Introduction
Diagnostic criteria of the syndrome of transient headache and Neurologic deficits with cerebrospinal fluid lymphocytosis, according to the International Headache Society (4).

HaNDL is most often seen during the third and fourth decades of life, although cases ranging from ages of 7–52 years have been reported, with no consistent gender predominance identified (3,5,6). Some patients report a preceding non-specific viral prodromal illness (6).
The etiopathogenesis of HaNDL is poorly understood. Studies have failed to provide a definite viral association. However, considering the presence of cerebrospinal fluid (CSF) pleocytosis, frequent infectious prodrome, and a monophasic course, a post-infectious or inflammatory mechanism in which autoantibodies directed against neuronal or vascular antigens has been proposed (2,3). The presence of antibodies directed against the CACNA1H subunit of the T-type voltage-gated calcium channel in one small study supported the inflammatory or autoimmune hypothesis (7). Similarly, even though only a minority of patients with HaNDL have a prior history of migraine or a family history of migraine (3,5), there are findings that are supportive of a migrainous pathophysiology suggested by single-photon emission computed tomography (SPECT) (8), perfusion imaging with computed tomography (CT) (9) and magnetic resonance imaging (MRI) (10), transcranial Doppler (11), and electrophysiology studies (5). A proposed unified hypothesis could be a post-infectious or inflammatory mechanism in which autoantibodies directed against neuronal or vascular antigens induce an aseptic leptomeningeal vasculitis and subsequent headache and neurologic deficits via a spreading depression-like mechanism (5,11,12).
Neurologic deficits typically last 15–120 minutes, although a range of 5 minutes to 3 days has been reported (2,3,5). Although approximately 75% of HaNDL cases have a monophasic course, repeated attacks of transient headache and neurologic deficits that occur for weeks or even months following the initial attack have been reported (2,3,5). The neurologic deficits often vary from one episode to the next, involving different brain regions. In the two largest reports, the mean durations of the illness were 14 and 21 days (2,3,5).
The neurologic manifestations (2,5) in HaNDL are protean and include hemi-sensory disturbances (70%), aphasia (66%),nausea/vomiting (54%),weakness (42%), visual complaints (including decreased vision, homonymous hemianopsia (18%), photopsia/photophobia (16%)), and rare reports of diffuse manifestations such as an acute confusional state (13). HaNDL may be accompanied by elevated intracranial pressure (ICP) and papilledema (14); however, a catastrophic presentation of impending cerebral herniation has not been previously described.
Herein, we describe two cases of HaNDL presenting as acute neurologic emergencies, which will reinforce and expand the spectrum of HaNDL-associated neurological manifestations.
Case report
Case 1
A previously healthy 31-year-old woman with no prior significant medical history except for obesity reported 2 weeks of progressive new-onset bi-frontal throbbing headaches with associated nausea, vomiting, photophobia/phonophobia and neck stiffness, but denied accompanying fever or systemic or constitutional symptoms. Sumatriptan and opioids provided no symptomatic relief. She subsequently developed double vision and pupillary asymmetry, which prompted evaluation at our institution, where her examination showed normal vital signs, intermittent anisocoria (left greater than right), bilateral papilledema (Figure 1B), and generalized hyper-reflexia with plantar flexor responses. After normal non-contrast brain CT, CT head and neck angiogram, and CT head venogram, a lumbar puncture (LP) showed an opening pressure (OP) >55 cm H2O, 390/cm3 white blood cells with lymphocytic predominance (94%), 463/cm3 red blood cells, protein 207 mg/dL (normal 15–60 mg/dL), and glucose 59 mg/dL (serum 112 mg/dL). The patient clinically improved and anisocoria resolved after withdrawal of a large volume of CSF by LP. MRI of the brain and of the entire spine with and without gadolinium contrast demonstrated diffuse leptomeningeal enhancement around the brain, but no other pathological findings (Figure 1A). Magnetic resonance venogram showed small left transverse, sigmoid, and internal jugular veins, but no evidence of thrombosis. She was empirically treated with acyclovir, vancomycin, ceftriaxone, and ampicillin, which were discontinued after negative bacterial and viral studies.
(a) Magnetic resonance imaging of the head with and without gadolinium (patient 1). Left: axial; right: coronal. No acute intracranial findings were noted; however, subtle post-gadolinium leptomeningeal enhancement was noted, and attributed to post-lumbar puncture changes. (b) Detailed fundus examination (patient 1) revealing prominent papilledema bilaterally with evidence of venous engorgement, blurring of the optic margins, and small hemorrhages adjacent to the optic disk, consistent with severe intracranial hypertension. (c) Computed tomography brain perfusion (patient 2). Upper left and upper right: cerebral blood volume and cerebral blood flow, respectively; lower right and lower left: mean transient time and time to peak, respectively. Notice the subtle asymmetries in the posterior left MCA territory (temporo-parietal region) with decreased cerebral blood flow, but increased time to peak, respectively, indicative of focal areas of brain hypoperfusion (while the patient was symptomatic). (d) Electroencephalography (patient 2) with focal slowing as demonstrated by polymorphic delta activity of moderate amplitude over the left temporal and frontal regions compared to the right hemisphere. MCA: Middle cerebral artery.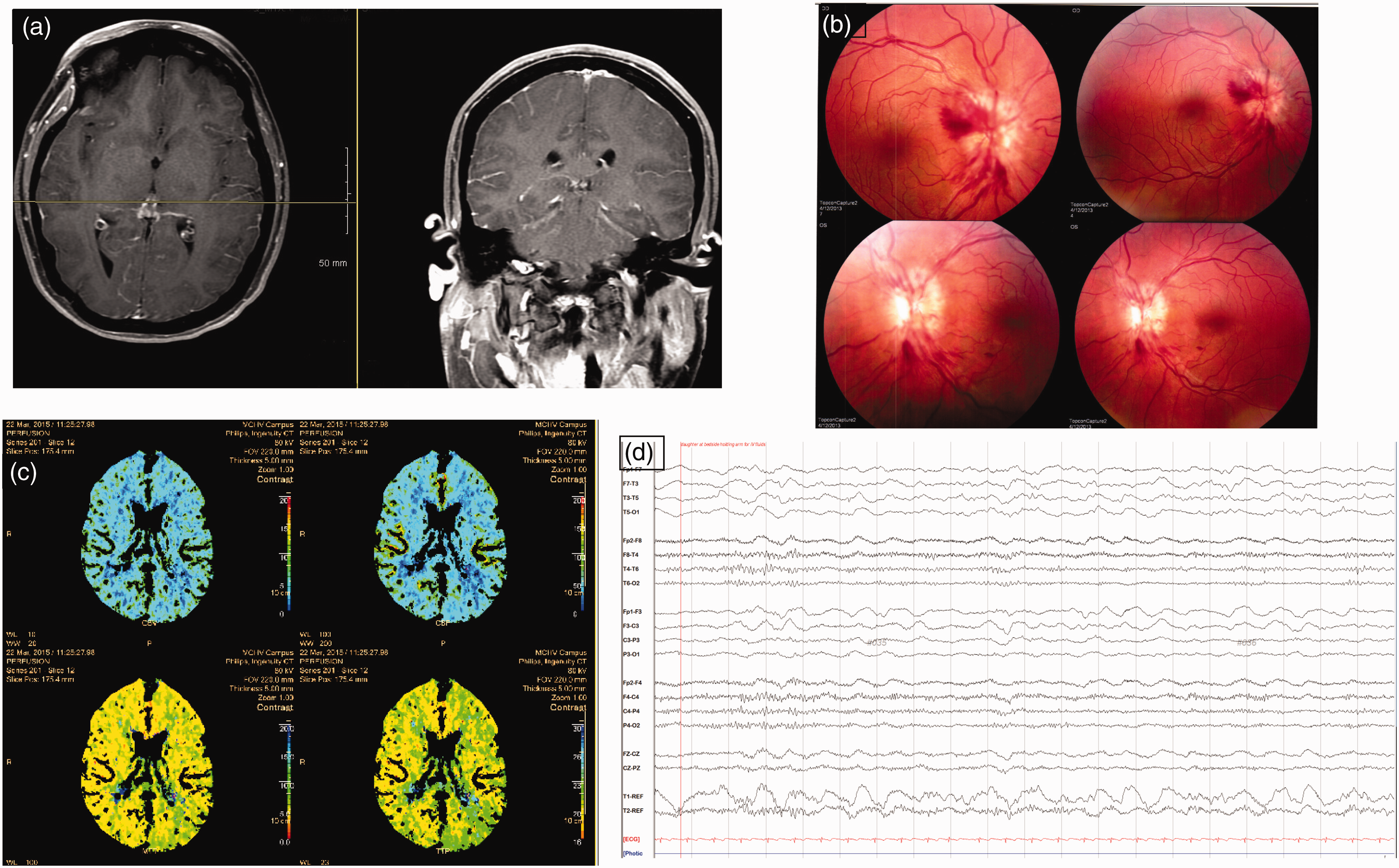
Three days after the LP, she again developed worsening of headache, fluctuating mental status, and findings suggestive of partial left third nerve palsy, prompting repeat LP, which showed OP >55 cm H2O, 625/cm3 white blood cells with lymphocyte predominance, and protein 325 mg/dL. Additional studies for fungi, tuberculosis, and CSF flow cytology were negative. A lumbar drain was placed and drained at 10 cc/hour for 3 days with symptomatic improvement. The lumbar drain was subsequently removed and, after observing for 3 days, the patient was discharged home with a presumptive diagnosis of aseptic meningitis causing elevated ICP.
She again presented to an outside hospital 3 days later with acute worsening of her headache and a change in mental status. Repeat LP again showed OP >55 cm H2O and persistent lymphocytic pleocytosis. While at the outside hospital, she was noted to have tonic extensor posturing and a rapid decline in mental status, prompting intubation for airway protection. She was also treated with levetiracetam and transferred back to our institution. After normal electroencephalography (EEG), levetiracetam was discontinued. She was extubated successfully the next day; a lumbar drain was placed again and repeated neuroimaging showed persistent diffuse leptomeningeal enhancement. Expanded evaluation was negative for infectious (rickettsial, mycobacterial, fungal, parasitic, and spirochetal) and autoimmune/inflammatory work-up (antinuclear antibody, anti-neutrophil cytoplasmic antibody, angiotensin-converting enzyme, rheumatoid factor, Anti-Sjögren's-syndrome-related antigen A/Anti-Sjögren's-syndrome-related antigen B (SSA/SSB), and paraneoplastic panel) and neoplastic (multiple flow cytometry and cytology) etiologies. Acetazolamide 500 mg twice daily was added and the lumbar drain removed after 1 week. She was subsequently discharged without any recurrence of symptoms. LP performed at 2 months and neuro-ophthalmology follow-up at 6 months post-discharge showed the resolution of CSF abnormalities and the resolution of prior papilledema that was present during the first admission, prompting discontinuation of acetazolamide. She remained symptom free without any recurrence of headaches at over 1 year of follow-up.
Case 2
A 68-year-old right-handed woman with a past medical history significant for episodic migraine without aura that was well controlled without any prophylactic medications and a new incidental diagnosis of prior well-healed (chronic) right carotid dissection presented with hyperacute-onset, mixed expressive and receptive aphasia and occipital headache of 4 hours’ duration. Headache was described as unilateral right sided, non-thunderclap but throbbing, and crescendo in nature, reaching its peak over 10–20 minutes, with associated phonophobia and photophobia, and was accompanied by new-onset aphasia.
The only preceding factor was vestibular exercises given for vestibular neuritis, which was diagnosed 2 weeks prior to the presentation. Work-up for vestibular neuritis then included MRI of the brain and magnetic resonance angiography of the head and neck vasculature, which revealed no acute findings apart from an old-appearing, well-healed right internal carotid dissection. Because of the lack of associated symptoms, this was thought to be an incidental finding and not related to this patient’s presentation.
On evaluation during the patient’s acute presentation, she was afebrile, had mild right nasolabial fold flattening, and mixed expressive and receptive aphasia. Brain CT perfusion revealed asymmetry of cerebral metabolism, with decreased cerebral blood flow and increased mean transient time in the posterior temporal and parietal lobes (Figure 1C), implying cerebral blood flow hypoperfusion in these areas. However, MRI of the head, which was performed twice over the next 3 days, was normal. CT angiogram of the head/neck showed widely patent intracranial and extracranial vasculature, except for the unchanged, previously described chronic right carotid dissection, suggestive of an incidental finding and not related to this acute presentation. LP showed OP of 26 cm H2O, 72 nucleated cells with 92% lymphocytic predominance, 73 red blood cells, and mildly elevated protein of 76 mg/dL. CSF studies for herpes simplex virus (I and II), herpes zoster virus, enterovirus virus, and bacterial culture all returned negative. EEG was performed (while the patient symptomatic), revealing polymorphic delta activity of moderate amplitude over the left temporal and frontal regions (Figure 1D). Continuous EEG monitoring failed to reveal any epileptiform or interictal epileptiform discharges. She was initially treated with acyclovir for herpes simplex, but this diagnosis was excluded by negative CSF studies and rapid clinical improvement following treatment with prochlorperazine, intravenous fluids, and magnesium. She had no sequelae of her initial presentation.
Discussion
HaNDL has previously been characterized as a self-limited headache syndrome with a benign course and for which therapeutic interventions other than symptomatic treatments are usually not needed. Our cases, while they confirm some of the previous observations, also provide additional features that contradict some of the previous assumptions.
Our first patient had findings suggestive of elevated ICP, supported by markedly elevated CSF OP >55cmH2O and clinical features including headache, bradycardia, intermittent third nerve palsy and subsequent obtundation with posturing, and loss of airway, which ultimately improved with large-volume CSF drainage. Although such a catastrophic presentation associated with HaNDL has not been reported before, in one study, elevated CSF OP was found in 56% of cases, ranging between 18 and 37 cm H2O (5). Moreover, papilledema has been present in multiple reports (3,5,14), and sixth nerve palsy and complete external ophthalmoplegia, which is related to effects of increased ICP, has been reported, which resolved with lowering of the ICP (14,15).
When evaluating a patient with headache, papilledema, and focal neurologic findings, a variety of disorders must be included in the differential diagnosis, including structural brain lesions, meningitis, seizures, and autoimmune, vasculitic and paraneoplastic disorders. Thus, a complete evaluation should be obtained when HaNDL is suspected, including imaging (MRI of the head, EEG, angiography, and venography) and serum/CSF studies for infectious, autoimmune, and vasculitic disorders. All of these etiologies were excluded in our patients. Idiopathic intracranial hypertension should also be considered, especially in obese patients who present with headache, papilledema, and increased ICP; however, neurologic deficits other than sixth nerve palsies coupled with an abnormal CSF profile make this diagnosis untenable (16).
Our first case also refutes the previous assumption that HaNDL is always a benign condition and that treatment of the syndrome consists essentially of symptomatic headache management. Even though our case eventually had a complete recovery, the potential for a devastating outcome existed had concomitant papilledema and raised ICP not been addressed aggressively. This conclusion is further supported by another report of HaNDL with papilledema and sixth nerve palsy, in which the patient progressed to optic atrophy, despite receiving acetazolamide and steroids (14). We suggest that the management of patients with HaNDL and elevated ICP, in addition to symptomatic headache treatment, should also include additional treatment with acetazolamide and, if required, temporary CSF drainage. Corticosteroids could also be considered (15).
Our first case confirms multiple rarely described previous observations. Despite HaNDL being considered a monophasic illness, clinical exacerbations may occur for weeks to months following the initial attack (3), hence the need for close surveillance and follow-up in suspected cases. CSF abnormalities may persist, despite clinical improvement, for an average of 99 days (range: 56–196 days) (17–19). In our first case, CSF abnormalities resolved over a period of 2 months. The observed leptomeningeal enhancement (18,19) might be explained on the basis of vasogenic leakage from the leptomeningeal vessels or be attributable to prior LPs. Such enhancement can also be seen in meningitis, brain tumors, status epilepticus, and stroke, which had been excluded by studies performed in our patient.
Our second case, presenting with occipital headache and aphasia, showed a normal CT scan and MRI of the head and an incidental finding of a stable, chronic, well-healed right internal carotid artery dissection, but focal cerebral hypoperfusion was documented by CT perfusion scan and an area of focal cerebral dysfunction over the left fronto-temporal region was identified on EEG, providing anatomical correlation of the neurological deficits.
Similar findings revealing global hemispheric or focal regions of hypoperfusion on CT or MRI perfusion techniques correlating with neurologic deficits in acute symptomatic patients with HaNDL have been described (9,10). Based upon additional reports involving SPECT (8), revealing a decreased radionuclide uptake over the cortical region corresponding to neurological deficits during the acute phase and transcranial Doppler sonography (11) showing vasomotor changes during attacks, it is hypothesized that hypoperfusion in HaNDL might be secondary to hypometabolism at the involved sites. Taken together, the changes in blood flow and associated EEG activity in our patient support the hypothesis that, similarly to migraine with aura, a HaNDL neurological attack may represent a wave of cortical spreading of depression with associated oligemia, possibly triggered by inflammation (5,11,12).
Conclusion
HaNDL, an enigmatic condition with diverse clinical manifestations, remains a diagnosis of exclusion. It may be explained on the basis of migrainous or post-viral/inflammatory pathophysiology. We describe two unusual presentations of HaNDL, which expand the spectrum of this disorder, and discuss therapeutic options. We alert treating physicians to recognize this rare disorder with its potential unusual manifestations.
Article highlights
Our first case expands the clinical spectrum of this disorder and refutes the previous assumption that headache and neurologic deficits with cerebrospinal fluid lymphocytosis (HaNDL) is always a benign condition. HaNDL cases presenting with concomitant papilledema and raised intracranial pressure need to be managed aggressively, which, in addition symptomatic headache treatment, also includes additional interventions such as temporary cerebrospinal fluid drainage, acetazolamide, and possibly corticosteroids. Despite HaNDL being considered a monophasic illness, clinical exacerbations may occur for weeks to months following the initial attack, hence the need for close surveillance and follow-up in suspected cases. Focal findings on cerebral perfusion studies and electroencephalography in our second case, with a presentation that was suspicious for acute cerebrovascular accident (CVA), support the hypothesis that, similarly to migraine with aura, a HaNDL neurological attack may represent a wave of cortical spreading of depression with associated oligemia, possibly triggered by inflammation.
Footnotes
Acknowledgements
We thank the patients’ families for their cooperation. We acknowledge the Department of Neurology at Duke University and the Department of Neurological Sciences at the University of Vermont for their continued support.
All four authors were responsible for the neurologic care and management of the patients described in this manuscript. All authors agree to the conditions outlined in the Authorship and Contributorship section of the information for authors. No statistical analysis was performed. Drs Applebee, Babi, Shapiro, and Waheed were involved in the drafting and revision of this manuscript.
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Shapiro serves on the Data Monitoring Committee for Lilly clinical trials. Dr Applebee reports that she is involved in clinical therapeutic trials sponsored by Novartis, Actelion, Genentech, and Opexa Therapeutics.
Ethical statement
This manuscript adheres to any relevant standards of reporting with a completed checklist where appropriate. Supporting data are available to researchers (upon request if needed).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.