
Abstract
Background
While the most accurate diagnosis of migraine typically requires a clinical interview guided by strict diagnostic criteria, an alternative approach that ascertains migraine by questionnaire in population-based settings has been instrumental in the discovery of common genetic variants influencing migraine risk. This result may be surprising. Population-based approaches are often criticized for limited ability to distinguish migraine from other forms of primary headache. It is thus useful to revisit prevailing ideas about population-based ascertainment of migraine to evaluate the extent to which this approach has potential for additional insights into migraine genetics and therefore pathophysiology.
Overview
We review recent findings suggesting that the success of the population-based approach is derived from the possibility of collecting much larger samples than in the clinic-based setting even at the risk of introducing phenotypic and genetic heterogeneity. The findings are also consistent with new appreciations for the genetic basis of many other common, complex clinical characteristics. However, clinic-based ascertainment and other settings will remain more effective than population-based approaches for investigating certain, often very specific aspects of migraine genetics.
Conclusion
We argue that the detailed genetic architecture of migraine, various aspects of methodology, and the ultimate sample size achieved by population-based ascertainment will be critical determinants of the future success of this approach to genetic analysis of migraine and its comorbidities.
Introduction
Although migraine, either with (MA) or without (MO) aura, has been documented as a discrete and common clinical entity for centuries dating back to antiquity (1), its presentation is remarkably diverse, so diverse that it has at times been viewed as a collection of syndromes (2). Historically, migraine has been distinguished by the presence of aura or the specific features of the migraine headache, most prominently its pain severity, duration, and its accompaniment with nausea and photophobia or phonophobia in the absence of other underlying etiologies, e.g. non-migraine disease. Diagnostic criteria for migraine have been formalized through the International Classification of Headache Disorders (ICHD), currently in its third version (3). In practice, the criteria are typically evaluated by a neurologist interview in a semi-structured way. Such a direct interview, either face to face in the clinic or by telephone, may elicit additional valuable information. For example, the interview may help establish the historical aspect of the diagnostic criteria requiring recurrence of headaches or differentiation from other headache types and neurological disorders. Furthermore, a direct interview affords the flexibility of characterizing premonitory symptoms as well as triggers, for example, which may be important with respect to advice on behavior and timing of treatment.
While the formalized diagnostic criteria for common migraine yield heritability estimates in the range 0.34–0.57 and suggest greater heritability for MA than MO (4–15), initial attempts to identify specific migraine risk loci by linkage analysis (16–27) and candidate gene association studies (28,29) were largely unsuccessful. This outcome contrasts with Mendelian forms of migraine exemplified by familial hemiplegic migraine, which has been successfully traced to channelopathies of monogenic origin (30). In the current era of genome-wide association studies (GWAS), we can now recognize that one explanation for the null findings for common migraine is likely a genetic architecture that has been observed for a majority of complex clinical traits (31). Instead of being explained by a handful of strong associations, much of the heritability of complex traits appears to be due to a vast number of weak associations at common genetic variants (e.g. minor allele frequency greater than the range 1–5%), although the contribution of rare variants is not fully understood and may differ according to trait (32,33). As a consequence, large samples are needed to achieve sufficient statistical power for detecting associations meeting robust significance thresholds for genome-wide analysis (34). However, even with a large sample, statistical power may be limited because of heterogeneity if cases of migraine vary with respect to underlying pathophysiology and genetics in spite of consistency with formal diagnostic criteria (33). The potential for such heterogeneity in migraine may be exaggerated compared with other complex clinical disorders (or conditions) precisely because its presentation is intrinsically so diverse, as reflected not only in the dichotomy of MA and MO forms, but also in the variety of migraine-associated traits, i.e. nausea, photophobia, phonophobia, unilateral pain. Nevertheless, while limiting analyses to groups with more homogeneous presentation might improve signal strength, it also would reduce sample size. Experience suggests that a better strategy for more power might be a truly dramatic increase in sample through ascertainment of migraine from populations, even if this approach risks introducing misclassification or heterogeneity.
Migraine ascertainment by questionnaire
The most efficient method for reaching large numbers of migraineurs in populations relies on self-reported responses to questionnaires developed, and in some cases validated, for assessing migraine. A questionnaire asking solely about an individual’s history of migraine will identify migraineurs with reasonable sensitivity but may also detect individuals with non-migraine headache (“false positives”), and fails to differentiate between MA and MO. More detailed questionnaires provide better consistency with the standard of clinic-based ICHD migraine classification by interrogating for critical diagnostic components such as the presence of aura, the frequency of headaches, and the characteristics of the headache. Consistency may be further enhanced by asking up to a total of 56 questions (35,36), for example about experience of prodromal symptoms, whether migraine has been previously diagnosed by a physician, the nature and habitual use of medications, and about potentially confounding conditions, all information that would be elicited in a physician diagnostic interview for affirming migraine as opposed non-migraine headache. Responses to these questions may then be used to classify individuals according to exclusively MA, exclusively MO, a combination of MA and MO, non-migraine headache (e.g. tension-type), medication-overuse headache, or non-headache controls. Evaluation of self-reported responses to this more thorough interrogation leads to quite substantial enrichment of migraineurs and exclusions of non-migraineurs. Using questionnaires, the consistency between self-report- and physician-based diagnoses can be quantified by the kappa statistic, a measure of inter-rater agreement, and is estimated in the range of 0.22–0.96 (35,37–41) with the higher values associated with more complex questionnaires. Similarly, the range of sensitivity for identifying migraineurs, i.e. the proportion of questionnaire-based migraineurs among those with physician-diagnosed migraine, is at least 0.5, while the range of specificity, i.e. the proportion of questionnaire-based non-migraineurs among those lacking migraine by physician diagnosis, is typically higher, even close to 1.0 for MA where responses to questions about aura are particularly diagnostic (35,40).
For genetic analyses, there are additional epidemiologic considerations about how a questionnaire designed to distinguish different forms of headache disorders is implemented and interpreted, and which populations are targeted for survey. Firstly, while questionnaire-based classifications may be used directly for establishing migraine, non-migraine headache, or controls, they may alternatively be used as a screening tool for follow-up by physician interview. This approach is expected to improve consistency with ICHD criteria compared with self-administered questionnaires alone while allowing access to much larger samples than may be available in the clinic (42).
Secondly, validation studies of migraine questionnaires often remark on a subset of “probable” migraineurs, who may claim to have experienced migraine yet do not meet full ICHD criteria although they may nonetheless nearly meet these criteria. Such cases are consistent with the notion of an underlying liability or more specifically genetic liability to migraine. In this framework, genetic risk is cast in terms of a (normally) distributed inherited genetic predisposition to migraine with somewhat arbitrary thresholds imposed to distinguish migraineurs from non-migraineurs, i.e. controls. Questionnaire-based probable migraineurs thus might be considered for inclusion as cases for genetic analysis (8,43).
Finally, regardless of how questionnaire responses are implemented or interpreted, population-based genetic findings will reflect characteristics of the targeted population, possibly in subtle ways. It may be obvious that questionnaires directed at youths may help identify genetic variants that are more relevant to migraine at an earlier age while targeting women may have potential to enrich for genetic variants on the X-chromosome or in biological pathways relevant to menstrual-related migraine. However, the implications of other population-specific designs may be less obvious. For example, if targeting women, studies focusing on female health care professionals, who are likely to report their health status more accurately than other population-based groups of women, may afford more statistical power. If combining samples from several sources in meta-analysis as is performed in GWAS, it remains an open question whether population-based ascertainment focusing on collections of families or twins with migraine probands, e.g. identified from registries of migraineurs, may harbor the genetic variation with the same prevalence and biological consequence as variation associated with migraine observed among unrelated individuals. Study designs that emphasize families for ascertainment of migraine, for example, may disproportionately enrich for rare migraine-associated variation rather than the common variants that would be identified by population-based ascertainment (44). However, discovery of the rare, migraine-associated variants by family-based analysis remains an essential component for understanding migraine pathophysiology since these variants are typically under-powered in the population-based setting and also may target genes that do not harbor common alleles for population-based analysis.
Genetics of population-based migraine
Population-based ascertainment of migraine in the IHGC GWAS (see Anttila et al. (45)).
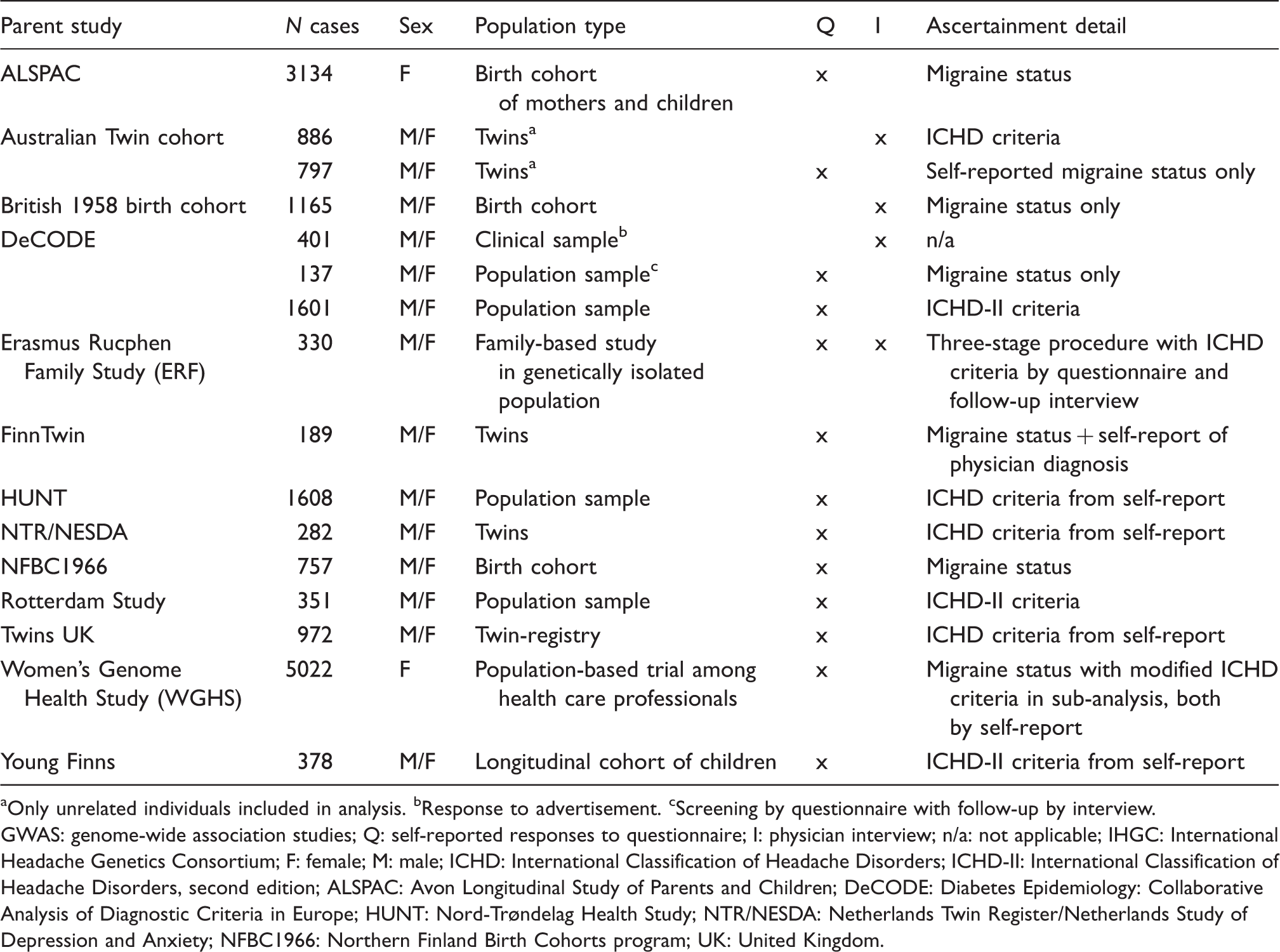
Only unrelated individuals included in analysis. bResponse to advertisement. cScreening by questionnaire with follow-up by interview.
GWAS: genome-wide association studies; Q: self-reported responses to questionnaire; I: physician interview; n/a: not applicable; IHGC: International Headache Genetics Consortium; F: female; M: male; ICHD: International Classification of Headache Disorders; ICHD-II: International Classification of Headache Disorders, second edition; ALSPAC: Avon Longitudinal Study of Parents and Children; DeCODE: Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe; HUNT: Nord-Trøndelag Health Study; NTR/NESDA: Netherlands Twin Register/Netherlands Study of Depression and Anxiety; NFBC1966: Northern Finland Birth Cohorts program; UK: United Kingdom.
Moreover, population-based ascertainment using questionnaires captures at least part of the diversity in migraine presentation precisely enough to demonstrate selective genetic associations correlated with phenotypic heterogeneity. In the WGHS, where the initial three migraine loci were identified, the study participants self-reported both migraine and headache status by questionnaire, providing a basis for defining a sub-group experiencing headache but not migraine headache (46). The association of the three loci with migraine was supported by the persistence of these associations with migraine compared to non-migraine headache (46). Similarly, in the WGHS, likelihood models demonstrated highly significant, selective associations between four of the 12 IHGC loci and migraine sub-classified according to characteristics relevant to ICHD diagnostic criteria (57) (Figure 1). These selective associations included an association at LRP1 with MO but not MA. Further, an association at a fifth locus, TRPM8, was significantly non-selective, perhaps consistent with the presumed role of this locus in the headache pain that is common to essentially all forms of migraine. These findings were subsequently supported by a similar investigation in a completely independent, clinically phenotyped collection of severe migraineurs from the Danish Headache Center and by analysis of heterogeneity in the original IHGC GWAS (56,58).
Selective association with sub-classified migraine from population-based ascertainment.
Although the 12 migraine loci identified by the IHGC from a largely population-based collection of migraineurs explain only a very small fraction of the heritability, the results are reminiscent of those from GWAS analysis of many other clinical traits. GWAS approaches identifying common variants meeting genome-wide significance have been most effective at accounting for heritability estimates from twin studies for clinical traits that have low phenotypic heterogeneity or little phenotypic ascertainment error and also low genetic heterogeneity or relatively simple genetic architecture (33,59,60). For example, a modest number of common specific loci identified for strongly genetic diseases such as type-1 diabetes (T1D) and age-related macular degeneration (AMD) explain a substantial proportion of the heritability or even most of the susceptibility (61–63). Similarly, known common loci explain much of the heritability in height, which is typically measured precisely, while most of the remainder can be formally attributed to other unknown but still common variants, although the height loci are far more numerous than those for T1D and AMD and each locus carries a relatively small effect (32,64,65). By contrast, for other clinical traits with more phenotypic and genetic complexity, known genome-wide significant common variants may explain only a fraction of the heritability (59). For example, coronary heart disease (CHD) and type-2 diabetes, which are thought to arise by a heterogeneous mix of etiologies, and blood pressure, which is difficult to measure precisely, have been intensively examined by GWAS incorporating very large samples without markedly explaining the estimated heritability, i.e. at most 10.6% and 5.7% of the heritability of CHD and type-2-diabetes, respectively, and 2.2% of the inter-individual variation in blood pressure (66–69).
Next steps
The question remains whether increasing samples of migraine cases in GWAS will continue to reveal additional migraine loci. The trajectory of discovered migraine loci with successively larger samples is consistent with the pattern observed for other heritable clinical traits, such that there is a linear trend in the yield of loci reaching genome-wide significance once a threshold sample size has been achieved (31). While, the number of migraine loci is fewer than for many other clinical traits with comparable heritability, it can be shown that moderate amounts of genetic heterogeneity, which may be introduced by misclassification of migraine in population-based ascertainment, may be partially overcome by dramatically inflating sample size (33,59). For example, for CHD or type-2 diabetes (66,68,70), it has been estimated that substantially explaining heritability would require an increase in sample size of 10-fold or more (60,71). For the case of CHD, it was recently demonstrated that more than doubling the number cases to 60,801 together with both more accurate and complete genotype information derived from an improved genotype imputation reference panel could increase the estimated proportion of heritability from approximately 11% to 25% (72).
The situation for migraine can be modeled explicitly in power calculations using effect estimates (i.e. odds ratios (ORs)) and minor allele frequencies consistent with the 12 published genome-wide significant loci (Figure 2). As much as 80% power may be reached for the largest effects (e.g. true OR = 1.25) in the presence of even 50% heterogeneity with sample size comparable to the previous IHGC GWAS (e.g. approximately 23,000 cases). However, much larger samples, even as large as 100,000 cases or more, are needed for detecting associations that may have smaller effects (e.g. true OR = 1.1) or that may be exclusive to MA compared with MO among migraineurs who have not been stratified by aura status or who are not exclusively MA, as may arise in population-based ascertainment. Looking to the future for potential sources of these needed additional samples, ongoing initiatives from the United Kingdom (UK) Biobank (73,74) or direct-to-consumer models for population-based ascertainment (75,76) will help, but the task may require even larger population samples and engagement organized at the national level of many countries (77), e.g. through questionnaire-based ascertainment methods.
Power for genetic association with migraine in the presence of heterogeneity.
The potential for continued success of population-based analysis may also be related to still unknown aspects of migraine’s genetic architecture. Others have called attention to analogies between migraine and a spectrum of neuropsychiatric disorders including schizophrenia, bipolar disorder, major depressive disorder, etc. (78). These conditions present with a diversity of symptoms, perhaps analogous to the diversity of aura status and headache features in migraine. While initial GWAS of the separate neuropsychiatric disorders revealed distinct loci for each, more recent analysis has emphasized appreciable genetic overlap, both at strongly associated, specific loci and in overall shared heritable determinants that include many weak genetic contributions across the genome (79,80). It is thought that some genetic variants, perhaps typically common variants, underlie shared aspects of the disease risk across the related disorders, but that other variants, including both common and rare, may be more specific for sub-classes of neuropsychiatric illness. Similarly for migraine, the heterogeneity in clinical presentation that nevertheless does not induce clear diagnostic sub-classification of migraine beyond MA versus MO may be due to a combination of shared susceptibility loci and loci conveying more specific susceptibility for MA versus MO or the features of the migraine headache (see also Figure 1).
Thus, because of either migraine’s genetic architecture or the difficulties in discerning potential migraine sub-classes beyond MA versus MO (if they exist) by current diagnostic techniques using questionnaires, population-based genetics may be most effective for identifying associations that are shared by all forms of migraine. A suitable analogy may compare population-based analysis of migraine genetics to a low-resolution photograph, in which many features of a scene are discernable, including many salient aspects, but some detail may be lost. Similarly, population-based genetic analysis of migraine may detect some of the strongest associations that are shared by the majority of migraineurs while having only modest potential to identify genetic correlates with sub-classes of migraine. Nevertheless, given how much remains unknown about the genetics and therefore pathophysiology of migraine, even partial clues from such “low-resolution” genetic associations advance our understanding. In the genomics era, when the aspirational goal is often complete knowledge, incomplete knowledge may still be quite informative (81) and may therefore advance thinking about therapeutic strategies.
The preceding discussion may be relevant to the lack of genome-wide significant associations for MA compared to a handful of associations for MO in a sub-analysis of the IHGC GWAS (45). Again, heterogeneity may supply part of an explanation (see also Figure 2). While establishing aura status by questionnaire is particularly reliable in its consistency with physician interview (35,40), it remains uncertain whether the general genetic architecture supports a distinction between MA and MO. A large proportion of the SNP associations in the analysis stratified by aura status, i.e. MA versus MO showed extensive and highly significant concordance in the direction of their effects on migraine of the two types, driven mostly by weakly associated SNPs, i.e. SNPs substantially below genome-wide significance (58). However, in the IHGC analyses of migraine stratified by aura status, a higher proportion of the cases were derived from clinic-based studies for MA (55%) compared with MO (33%). Although other explanations are possible, it may be that aura presentation tends to have more genetic heterogeneity that may have been accentuated by the higher proportion of cases from clinic-based ascertainment in the IHGC. That is, in the IHGC samples, MA status may have been due to some common alleles from the population-based studies but distinct rare alleles from the clinic-based studies. This potential source of genetic heterogeneity, and possibly others that are not recognized, may have limited the identification of genome-wide significant associations with MA in the meta-analysis. It is not known whether similar discrepancy of MA versus MO loci would have been seen with exclusively population-based ascertainment using the standardized complex questionnaires designed to recapitulate aspects of an ICHD-based diagnostic interview. Genetic association with MA but not MO may also be revealed only in a setting in which individuals who experience solely MA or solely MO are compared. Notably, distinguishing those migraineurs experiencing solely MA or MO from those experiencing both is particularly challenging using questionnaire-based ascertainment (35,40).
Comparison to genetics of migraine ascertained in other settings
The paucity of MA compared with MO loci further highlights why ascertainment in the clinic or other unique settings as opposed to the population may represent a more efficient strategy for detecting genetic associations with specific migraine presentations. Clinic-based migraine is likely to be more severe and often may reflect historical features of a local population or a family structure, such that relevant migraine-associated variants may be unique or more penetrant than variants for common migraine. Regarding the former, the per-allele effect estimates of the 12 IHGC GWAS SNPs were stronger in the clinic-based samples than in the population-based samples (45,58). Similarly, one of the five novel SNPs in this study was discovered only in analysis of the samples with exclusively clinic-based ascertainment. Regarding the latter, some of these same clinic-based samples had been examined previously in the first GWAS of migraine to identify and replicate a SNP at chromosomal location 8q22.1 with primarily MA association (48). This SNP, rs1835740, was not consistently replicated in other independent clinic- or population-based samples (46,49,54,82,83) and therefore may have population-specific properties. Similarly, a pedigree from the clinic with circadian rhythm abnormalities and associated MA or MO was resolved by sequencing to a novel rare variant in the ubiquitously expressed casein kinase 1δ (CK1δ) gene that was validated by a second variant in an independent pedigree (84). Neither variant is observed in population-based samples. Another unusual cohort of migraineurs consists of the inbred Norfolk Island descendants of the Bounty, whose complex pedigree has an exceedingly high prevalence of migraine of almost 26%. A candidate migraine variant on the X-chromosome from this pedigree was supported in a population-based sample of common migraine (85,86). As in the history of human genetics, these examples illustrate the potential for unusual ascertainment scenarios focused around the clinic for discovery of migraine associations that may not have been found by a population-based analysis but nevertheless may still provide insights into migraine pathophysiology.
Additional aspects of population-based migraine genetics
Genome-wide genetic data that are the basis for GWAS may also be used to estimate the genetic correlation (also known as bivariate heritability) between migraine and its comorbidities. These analyses address the extent to which the genetic determinants of migraine are shared with its comorbid conditions, such as ischemic stroke, CHD, epilepsy, restless legs syndrome (RLS), depression, or cervical artery dissection (87). Recent statistical techniques further allow partitioning shared genetic contributions according to regions of the genome with identifiable biological functions, e.g. supporting gene expression for specific biological pathways or in particular cell types, e.g. vascular or neurological cell types (88,89). Because shared genetics implies shared biological properties, such information may be useful in delineating the shared pathophysiology underlying migraine and its comorbidities. However, this type of analysis is subject to the same issues of population versus clinic ascertainment that influence the outcome of genetic analysis of migraine alone or the comparison of MA and MO as described above. Namely, when possible, population-based ascertainment of the migraine comorbidities is likely a better route to large sample sizes required for statistical power but will most efficiently capture genetic influences that underlie the broadest aspects of etiology. For example, CHD ascertained in populations might capture genetics that are shared among events that are aggregated in the definition of CHD, e.g. need for revascularization and myocardial infarction. By contrast, clinic-based ascertainment might be a better approach for reliably distinguishing genetic influences that may be unique to either of these two forms of CHD. Other migraine comorbidities such as ischemic stroke, epilepsy, and depression are believed to be even more heterogeneous than CHD; and in fact, neither GWAS of clinic-based ischemic stroke nor bivariate heritability analysis of migraine and stroke has revealed shared genetic influences that are thought to exist (90). For the comorbidities that are relatively common, e.g. prevalence of at least 1%, population-based ascertainment strategies are typically available through questionnaire (e.g. RLS (91–96)) or the combination of screening by questionnaire followed by physician review of medical records (CHD, ischemic stroke, depression), e.g. Ridker et al. (97). However, for rarer comorbidities, such as cervical artery dissection with incidence estimated at 2.6/100,000/year (98,99), clinic-based ascertainment may be the only viable option.
Epidemiological considerations support one additional application for population-based ascertainment in genetic analysis of migraine, namely the potential for discovery of environmental exposures that interact with genetics to modify migraine risk. A major theme of migraine research with clear translational implications is the identification and mechanistic inquiry into exposures that trigger migraine attacks (100–102). In theory, exposures that may be protective are also possible (103). Whether effects of these environmental influences may be modified by genetics is unknown, but robust investigation should be optimally performed in the population-based setting. The analytic challenges due to heterogeneity and weak effects observed for detection of main effects in genetics are expected to be amplified for gene-by-environment interaction and would likely be overcome only with large sample sizes available by population-based ascertainment. Moreover, while the case-only design for detecting gene-by-environment interactions could be performed in clinic-based studies in principle, this approach is predicated on the assumption of independence between the genetic marker and the exposure in the population from which the migraineurs are drawn (104,105). This assumption would be nearly impossible to verify in clinic-based samples, although their more accurate phenotyping may have value in identifying candidate gene-by-exposure combinations for follow-up in population-based studies.
Conclusions
However imperfect, ascertainment of common migraine in population-based settings through questionnaire or other assessment tools has been an essential element of the recent but long overdue successes in migraine genetic analysis. The experience with migraine follows the empirical paradigm observed for genetic analysis of other complex traits for which associations have been identified by prioritizing increased sample size over homogeneous phenotyping. As a consequence, these studies target the typically common genetic variation that is shared across phenotypic sub-classes. For migraine, clinical and genetic heterogeneity may be particularly complex. Genetic studies of migraine have yet to account appreciably for the higher heritability of MA compared with MO, and similarly do not yet resolve the genetic basis of the variation in features that accompany the migraine attack. These shortcomings are partly due to lack of studies designed to address these specific goals, but recent results suggest that questionnaire-based ascertainment may be adequate for such inquiry. With ever increasing, population-based samples for genetic analysis of clinically important traits, including migraine, now a central public health research priority in many countries (106), one may anticipate more success of migraine genetic analysis toward the essential goals of resolving the heterogeneity in migraine presentation, facilitating greater precision in the diagnosis of migraine suffers, and elucidating the pathophysiologic basis of migraine for the development of therapeutic strategies.
Key findings
Population-based samples have been essential to recent progress in understanding genetics of common migraine and should continue to be included in future analysis. The success of population-based ascertainment for genetic analysis migraine reflects both the possibility of collecting very large samples and the genetic architecture of complex clinical traits in general. However, limitations in the accuracy of population-based ascertainment of migraine may imply that it will likely need to be supplemented by other ascertainment methods, especially clinic-based ascertainment, for complete genetic dissection of common migraine.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.