
Abstract
Background
Migraine patients develop attacks several hours after intravenous infusion of glyceryl trinitrate. Due to the short half-life of nitric oxide, this delayed migraine cannot be caused by a direct action of nitric oxide derived from glyceryl trinitrate. The involvement of meningeal inflammation and dural mast cell degranulation is supported by the effectiveness of prednisolone on glyceryl trinitrate-induced delayed headache.
Methods
Using a newly developed rat model mimicking the human glyceryl trinitrate headache model, we have investigated the occurrence of dural mast cell degranulation after a clinically relevant dose of glyceryl trinitrate.
Results
A 6-fold increase in degranulation was observed starting at 2 hours after glyceryl trinitrate infusion. Interestingly, pre-treatment with the effective anti-migraine substances L-nitro-arginine methyl ester and sumatriptan prevented glyceryl trinitrate-induced mast cell degranulation whereas the calcitonin gene-related peptide-receptor antagonist olcegepant and the substance P receptor antagonist L-733,060 did not affect mast cell degranulation. However, topical application of two different nitric oxide donors did not cause mast cell degranulation ex vivo.
Conclusions
Direct application of an exogenous nitric oxide donor on dural mast cells does not cause mast cell degranulation ex vivo. In vivo application of the nitric oxide donor glyceryl trinitrate leads to a prominent level of degranulation via a yet unknown mechanism. This effect can be completely blocked by inhibition of the endogenous nitric oxide production and by 5-HT1B/1D receptor agonists but is unaffected by calcitonin gene-related peptide and substance P receptor antagonists.
Keywords
Introduction
In both humans (1,2) and rodents (3–5), the dura mater is densely populated with mast cells that reside in proximity to blood vessels and meningeal nociceptive fibres (4). Mast cells are closely related to basophile granulocytes and are part of the immune system. They mature in the tissue, where they are embedded (4). Mast cell degranulation can either be mediated through binding to immunoglobulin E (IgE) via the high-affinity receptor FcɛRI or can be IgE independent and initiated by neuroactive compounds, including sensory neuropeptides such as calcitonin gene-related peptide (CGRP), vasoactive intestinal polypeptide, substance P and pituitary adenylate cyclase activating peptide (6–8). Administration of the basic secretagogue compound 48/80 causes a prolonged mast cell degranulation and excitation of meningeal nociceptors that is accompanied by an increase of the phosphorylated form of extracellular signal-regulated kinase (p-ERK) in CGRP containing dural nerve fibres. Hence, degranulation of dural mast cells can lead to activation of dural nociceptors stimulating the headache pain pathway (9).
Infusion of glyceryl trinitrate (GTN), a donor of nitric oxide (NO), induces immediate headache in humans, which 5–6 hours later in migraine patients is followed by a delayed headache indistinguishable from a spontaneous migraine attack (10–12). The role of NO in the pathogenesis of migraine is further evidenced by the alleviation of migraine pain through inhibition of endogenous NO production by the non-selective nitric oxide synthase (NOS) inhibitor L-NG-monomethyl arginine citrate (13). Moreover, up-regulation of inducible NOS (iNOS) and inflammatory markers together with a concomitant degranulation of mast cells has also been observed in dura mater of rats following GTN infusion (14). Involvement of pro-inflammatory substances was suggested as the anti-inflammatory drug, prednisolone, reduced the intensity of the delayed headache in migraine patients after GTN infusion (15). We have recently developed an experimental GTN-induced headache model using GTN infusion in unanaesthetized freely moving rats. It is the closest possible simulation of the human GTN model of migraine. Our model circumvents confounding factors like high doses of GTN, anaesthetics, acute surgical stress and hypotensive effects associated with earlier GTN infusion studies in animals (16). Using this model, we found increased expression of c-fos and immunoreactivity of CGRP in laminae I/II of the trigeminal nucleus caudalis, c-fos indicates the activation of spinal trigeminal neurons. CGRP and neuronal nitric oxide synthase (nNOS) immunoreactivity were increased in dura mater (Figure 1). Pre-treatment with sumatriptan, L-nitro-arginine methyl ester (L-NAME), olcegepant and L-733,060 significantly decreased the GTN-induced c-fos expression in this model (16,17). NO acts as a retrograde signalling molecule and was shown to release CGRP from dura mater encephali (18). The release of trigeminovascular sensory neuropeptides may be inhibited by sumatriptan (19). We hypothesized that NO causes mast cell degranulation via peripheral activation of sensory neurons. To test this hypothesis, we used our validated rat GTN provocation model investigating the effect of L-NAME, sumatriptan, olcegepant and L-733,060 on mast cell degranulation in dura mater following GTN infusion. A second hypothesis, that NO donated from GTN is not directly responsible for mast cell degranulation, was also addressed in this study.
A schematic overview of changes in the rat dura mater 4 hours after infusion of GTN (4 µg/kg/min, for 20 min). The drawing shows nNOS immunoreactivity (brown) and CGRP-immunoreactivity (red) containing nerve fibres in dura mater in (a) vehicle and (b) GTN-treated rats (16). In the drawing, intact and degranulated mast cells are shown close to the middle meningeal artery (MMA) as found in the present study. Please note that when drawing this figure we have not taken into account the possibility of nNOS and CGRP being co-localized in some of the nerve fibres. We have also not been looking into the localization of nerve fibres in relation to each other, the MMA or mast cells.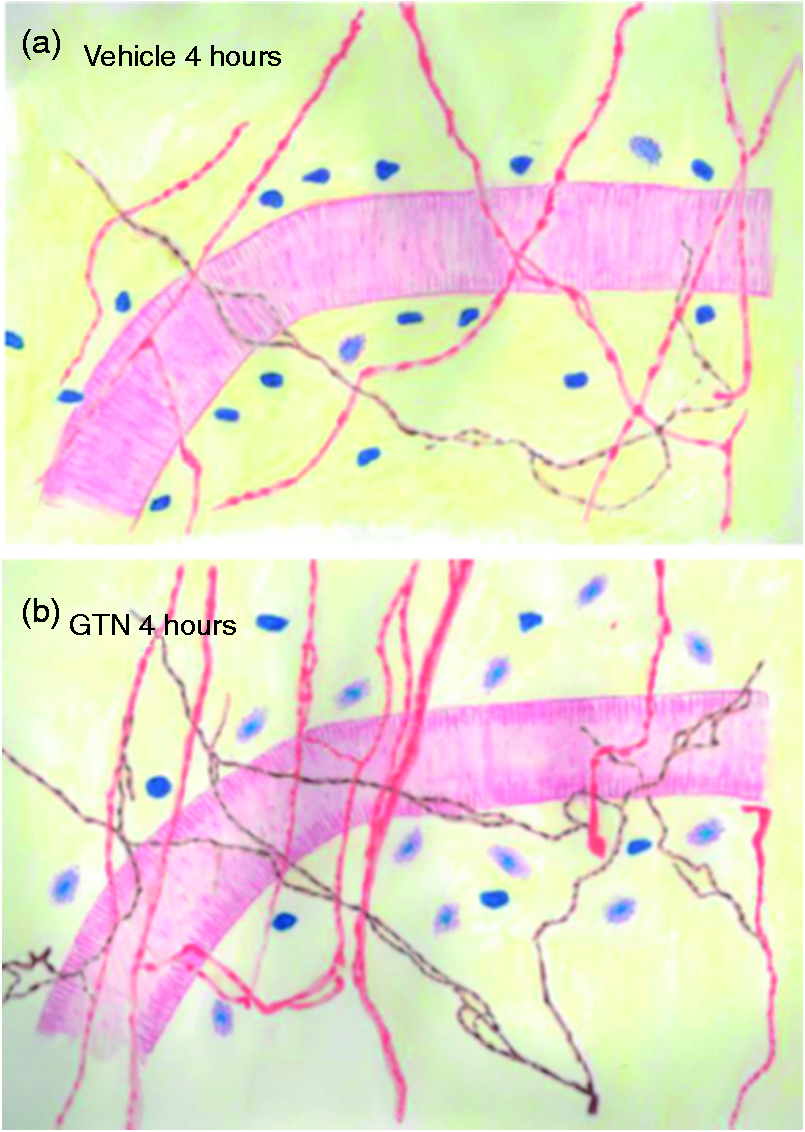
Materials and methods
Animals
A total of 94 male Sprague–Dawley rats weighing 250–320 g (Taconic M&B, Denmark) at the time of infusion, were used for this study. The rats were maintained in cages with a 12 hour light/dark cycle and given free access to a standard rodent diet and water at all times. Subsequent to surgery, animals used for in vivo experiments were single housed, whereas animals used for ex vivo experiments were group housed. All experimental protocols were approved by the Danish Animal Experiments Inspectorate (licence number 2012-15-2934-00697).
Surgical procedure
The surgical procedure was performed with modifications as described previously (16). Rats were anaesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg, Intervet, Skovlunde, Denmark) and xylazine (7.5 mg/kg, Rompun, Bayer Inc., Germany). The animals were monitored for depth of anaesthesia and deprivation of pain reflexes was regularly confirmed throughout the surgical procedure by toe pinch and evaluation of respiratory rates. Core body temperature was monitored and maintained using an automatic regulated heating pad (Letica HB101, Panlab, Barcelona, Spain). Eyes were covered with Viscotears® eye gel (Novartis Healthcare, Copenhagen, Denmark). Before surgery the rats were subcutaneously (s.c.) injected with the anti-inflammatory and bacterial drugs enrofloxacin (10 mg/kg, Rompun) and carprofen (5 mg/kg, Pfizer, NY, USA). Surgery was performed aseptically. The femoral or jugular vein was cannulated using polyethylene tubing (0.40 mm, ID 0.80 mm OD, Portex®, Smiths Medical ASD, USA). The catheter was placed s.c. and exteriorized at the nape of the neck. It was then checked for patency and sealed with heparinized saline (Heparin 500 IU/ml, Glostrup Hospital Pharmacy, Denmark). The rats were monitored until fully recovered from anaesthesia. Post surgery, they were given three doses of enrofloxacin and carprofen s.c. with 24 hour intervals and two doses of buprenorphine (0.03 mg/kg, Schering Plough, Belgium). They were then allowed to recover for a period of 7 days while the catheter was flushed regularly with heparinized saline to maintain patency. The animals were transferred to accusampler cages (Dilab, Lund, Sweden) and the catheter was connected through a tether. In this set-up they acclimatized for another 2 days while they were able to move freely within the cage.
Time course study of mast cell degranulation after GTN infusion
In order to determine the time-point where the effect of GTN reaches a stable level of mast cell degranulation, a time course study was performed. On post-surgical day nine the rats were subjected to i.v. administration of GTN (5 mg/ml in 95% ethanol, Nycomed, Roskilde, Denmark) in a dose of 4 µg/kg/min for 20 min and subsequently sacrificed at different time points (30 min, 2 hours, 4 hours or 6 hours, n = 3–5). Control animals were given vehicle (1.2% ethanol in saline) and sampled after 30 min and 2 hours (n = 3–4).
Pre-treatment with migraine-relevant substances in vivo
The effect of pre-treatment with migraine relevant substances on dural mast cell degranulation was studied. L-NAME (40 mg/kg, n = 8) was infused over 10 min and 5 min later followed by GTN. Sumatriptan (0.6 mg/kg, n = 7, Imigran®, Glostrup Hospital Pharmacy, Denmark) was infused over 3 min and 5 min later followed by GTN. Ten min prior to GTN infusion, olcegepant (1 mg/kg, n = 7, Tocris Bioscience, Bristol, UK) or the NK1-antagonist, L-733,060 (1 mg/kg, n = 7, Tocris Bioscience) was infused for 3 min. In control and vehicle groups, eight and nine animals were included, respectively.
For in vivo infusion, all stock solutions were diluted in sterile saline to their final concentrations just before administration. Animals from all groups were randomized on all experimental days. Pre-treatment with all four migraine-relevant substances was conducted as part of the same study and the vehicle- and GTN-treated groups are therefore the same throughout this study. Four hours after infusion, the rats were deeply anaesthetized in pentobarbital (Glostrup Hospital Pharmacy, Denmark) and euthanized by transcardial perfusion with phosphate buffered saline (PBS) followed by tissue fixation (4% paraformaldehyde (PFA) in PBS (Sigma-Aldrich, Schnelldorf, Germany). A craniotomy was made and the skull was cut mid-sagittal. The brain halves were removed and dura mater was sampled for histological analysis.
Dural mast cell degranulation ex vivo
The rats were sedated by inhalation of a CO2/O2 mixture followed by CO2 asphyxiation and decapitation. The skull was cut mid-sagittal and the brain halves were removed, taking great care to leave the dura mater undisturbed while attached to the skull. The skull halves were placed in concave clay moulds lined with Vaseline to prevent leakage immediately followed by the addition of 300 µl test substance. Either GTN (1 µM, n = 5) or sodium nitroprusside (SNP, 100 µM, n = 5, Sigma-Aldrich) was added in one skull half while the other skull half was subjected to PBS to serve as paired control in order to reduce experimental and biological variation. Compound 48/80 (1 µM, n = 8, Sigma-Aldrich) was used as a positive control for degranulation. Sumatriptan (30 µM) was added to both skull halves and incubated for 10 min prior to administration of either PBS or compound 48/80. The skulls were incubated for 10 min in a humid incubator at 37℃. The test substances were removed and skull halves were post-fixed in 4% PFA.
Histology
To identify the degranulation status in dural mast cells, we used Toluidine Blue staining, which visualizes mast cells by staining the heparin located in their cytoplasmic granules. Completely degranulated mast cells are not visible in this assay and may therefore not be detected. For both in vivo and ex vivo studies, the dura mater was dissected from the skull halves in whole pieces by carefully separating it from the skull with a spatula and cutting around the trigeminal ganglion. Dura mater was mounted flat on slides and stained with acidified 0.1% Toluidine Blue (Sigma-Aldrich). The tissues were dehydrated in graded alcohols and cleared in xylene prior to cover slip mounting. Mast cells were visualized under 400 ×magnification and were counted in 10 consecutive visual fields along the stem part of the middle meningeal artery (Figure 2(a)). Intact mast cells appear as intense dark purple cells (Figure 2(b)) and were considered degranulated if there was an extensive dispersion of more than 10 extruded granules localized near the cell or when there was an extensive loss of staining resulting in a ‘ghostly look’ (Figure 2(c)) as described by Levy and colleagues (9). The degree of degranulation is given as percentage of total number of counted mast cells. Counting was performed by a person blinded to the treatment.
Toluidine Blue stained rat dura mater whole mount. (a) Mast cells appear as densely stained purple cells located with a high density close to the vasculature. The Y-shaped area surrounding the middle meningeal artery (MMA) and its first branch designates the area in which mast cells are quantified in 10 consecutive fields. (b) and (c) illustrate the differences between intact and degranulated dural mast cells. Dura mater was incubated ex vivo with either (b) PBS as a baseline control (vehicle) or (c) 1 µM compound 48/80 and incubated for 10 min. Scale bar indicates 100 µm.
Wire myograph experiments
Rat brains from the ex vivo mast cell degranulation studies were immediately placed in cold Krebs buffer as previously described (20). In short, using a dissection microscope, segments of 1–1.5 mm in length of the middle cerebral artery was carefully isolated and mounted in the myograph (Multi Myograph System 610 M, Danish Myo Technology, Aarhus, Denmark) immediately after isolation. Two stainless steel wires with a diameter of 25 µm were carefully inserted in the lumen of the segments and attached to the jaws of the myograph bath. The measured isometric forces were digitized and transferred to a computer running data acquisition software (Myodaq, Danish Myo Technology). In order to achieve maximal force development, the vessels were normalized to a transmural tension of 13.3 kPa (L100). The vessels were set to the internal circumference L1 = 0.9 x L100. The buffer was continuously bubbled with 5% CO2 in O2, oxygenating the buffer and providing a pH of 7.4. The set-up was temperature controlled and kept at 37℃.
After normalization, the segments were allowed to rest for 1 hour, after which the buffer in the tissue baths was substituted with a 60 mM K+-rich buffer initiating a contraction, which was taken as a measure of contractibility of each segment. The contraction amounted to 0.43 ± 0.10 mN/mm (n = 8). In order to study the bioavailability of NO from the donors, GTN and SNP, the arteries were pre-contracted with the 60 mM K+-buffer that resulted in a stable tension of 0.47 ± 0.04 mN (n = 24). A cumulative concentration–response (C-R) curve was established in the concentration range of 10−8–10−4 M of freshly prepared drug. Each addition was at least 3min apart. After completing the C-R curve, the vessels were washed in Krebs buffer and once again contracted with 60 mM KCl buffer. The direct dilatory effect of NO on the vessels was validated by pre-treatment with a guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, Tocris Bioscience) that was added to the tissue baths 10 min prior to addition of the NO donor.
For the wire myograph, experiments the changes in contraction, measured in mN per mm vessel length, are expressed as percentage relaxation of the force measured on the vessel at the stable level of tension of the pre-contraction. All concentrations given represent the final concentration in the wire myograph tissue bath.
Statistical analysis
All in vivo and ex vivo skull model data were analysed by one-way analysis of variance (ANOVA) followed by pair-wise comparisons using Bonferroni’s multiple comparisons test or Dunnett’s multiple comparison test in case of significant effects. Myograph data were analysed by Mann–Whitney U test. Differences were considered statistically significant when p < 0.05 and data are represented as means ± SEM. GraphPad Prism (GraphPad Prism software, San Diego, CA, USA) was used for statistical analysis.
Results
Time-course effect of GTN-induced mast cell degranulation in vivo
The basal level of mast cell degranulation in vivo was determined 30 min and 2 hours after a 20 min infusion of vehicle to awake freely moving rats. The level of degranulation was 9 ± 1% (n = 3) at the 30 min time-point and stayed at 9 ± 2% (n = 4) for at least 2 hours after the vehicle infusion. Thirty min after GTN infusion, mast cell degranulation was increased by 2-fold to 20 ± 3% (n = 3). Two hours following GTN infusion, a significant 6-fold increase of 61 ± 15% (n = 5) degranulation was observed compared with controls. This level was maintained for 4 hours (66 ± 6 %, n = 5) and 6 hours (55 ± 9 %, n = 4) following GTN infusion (p = 0.0042, Figure 3). Based on these results, the 4-hour time-point was selected for subsequent investigation of the effect of migraine-relevant drugs.
Time-course study of GTN induced mast cell (MC) degranulation after GTN infusion (4 µg/kg/min for 20 min) in vivo. The level of degranulated MCs is given as percentage of the total number of mast cells in the counted area. No differences were observed between 30 min and 2 hours after vehicle (veh) infusion. However, an overall effect of GTN 2, 4 and 6 hours after infusion was observed (one-way ANOVA, p = 0.0042). *Represents p < 0.05 and denotes significant difference from 2 hours veh as detected by Bonferroni's multiple comparison test. Values are given as mean ± SEM, n = 3–5.
Effect of L-NAME on GTN-induced mast cell degranulation in vivo
Pre-treatment with the non-specific NOS inhibitor L-NAME (40 mg/kg) prior to GTN infusion abolished the degranulatory effect of GTN 4 hours after infusion. The level of degranulation was significantly reduced from 52 ± 8% (n = 9) to 12 ± 2% (n = 8), a level that did not differ from the basal degranulation of 19 ± 6% (n = 8, p = 0.0002) (Figure 4).
Effect of L-NAME on GTN induced dural mast cell (MC) degranulation. GTN-induced dural MC degranulation in L-NAME pre-treated animals in vivo. The level of degranulation is given as percentage of degranulated cells of the total number of counted mast cells. Vehicle and L-NAME pre-treated animals did not show any significant difference. # Represents significant difference from vehicle infusion whereas * represents significant difference from GTN treatment (one-way ANOVA, p = 0.0002, Bonferroni’s multiple comparisons test). Please note that the vehicle and GTN treated groups are the same as in Figures 7 and 9. Values are given as mean ± SEM, n = 8–9.
Ex vivo effect of GTN and SNP on mast cell degranulation and vasodilation
We applied a NO-donor topically to the dura mater in an ex vivo design. The basal level of mast cell degranulation was determined after incubation with standard isotonic PBS. Neither GTN (1 µM) nor SNP (100 µM) gave rise to degranulation after 10 min as the degranulation levels remained at 11 ± 1% and 15 ± 3% as compared with paired baseline levels of 15 ± 4% and 12 ± 2%, respectively (n = 5, p = 0.6149, Figure 5).
Ex vivo effect of NO donated from either GTN or SNP on dural mast cell (MC) degranulation. Intact and degranulated MCs were quantified and degranulation levels are given as percentage of degranulated cells of the total number of counted mast cells. Neither of the NO donors GTN (1 µM) or SNP (100 µM) gave rise to any significant degranulation as compared with vehicle (one-way ANOVA, p = 0.6149). Values are given as mean ± SEM, n = 5.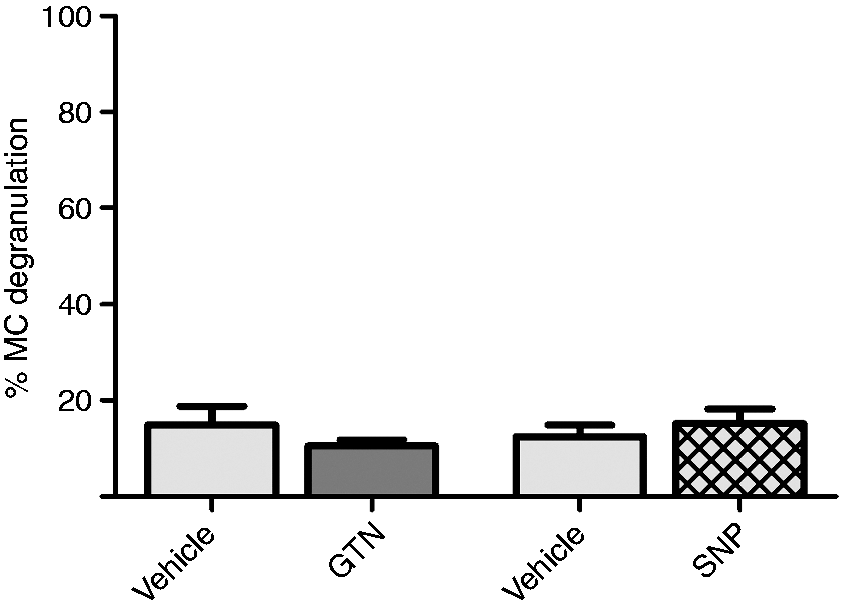
The actual release of NO from GTN and SNP ex vivo was confirmed by wire myograph studies performed on rat middle cerebral arteries. Single doses of GTN (1 and 10 µM) induced relaxant effects of 40 ± 7% (n = 4) and 68 ± 1% (n = 4), respectively of the potassium induced pre-contraction (Figure 6(a,b)). The same was true for a 100 µM SNP administration resulting in a 74 ± 3% (n = 4) relaxation of the artery (Figure 6(c,d)). For both GTN and SNP, vasodilation was completely blocked by pre-treatment with 10 µM of ODQ 10 min prior to GTN or SNP administration (Figure 6(b,c)).
Relaxant effect of NO on cerebral arteries ex vivo. (a) and (c) show representative traces of wire myograph experiments on middle cerebral arteries. The vessels were pre-contracted with 60 mM KCl-buffer to each tissue bath and when the contraction was stabilized (10–15 min) an NO donor was added: (a) 10 µM GTN or (b) 100 µM SNP. (b) and (d) summarize the relaxant effects of GTN (10 µM) and SNP (100 µM), respectively, in the presence and absence of ODQ (10 µM). * Represents p < 0.05, Mann–Whitney U test. Values are given as mean ± SEM, n = 4.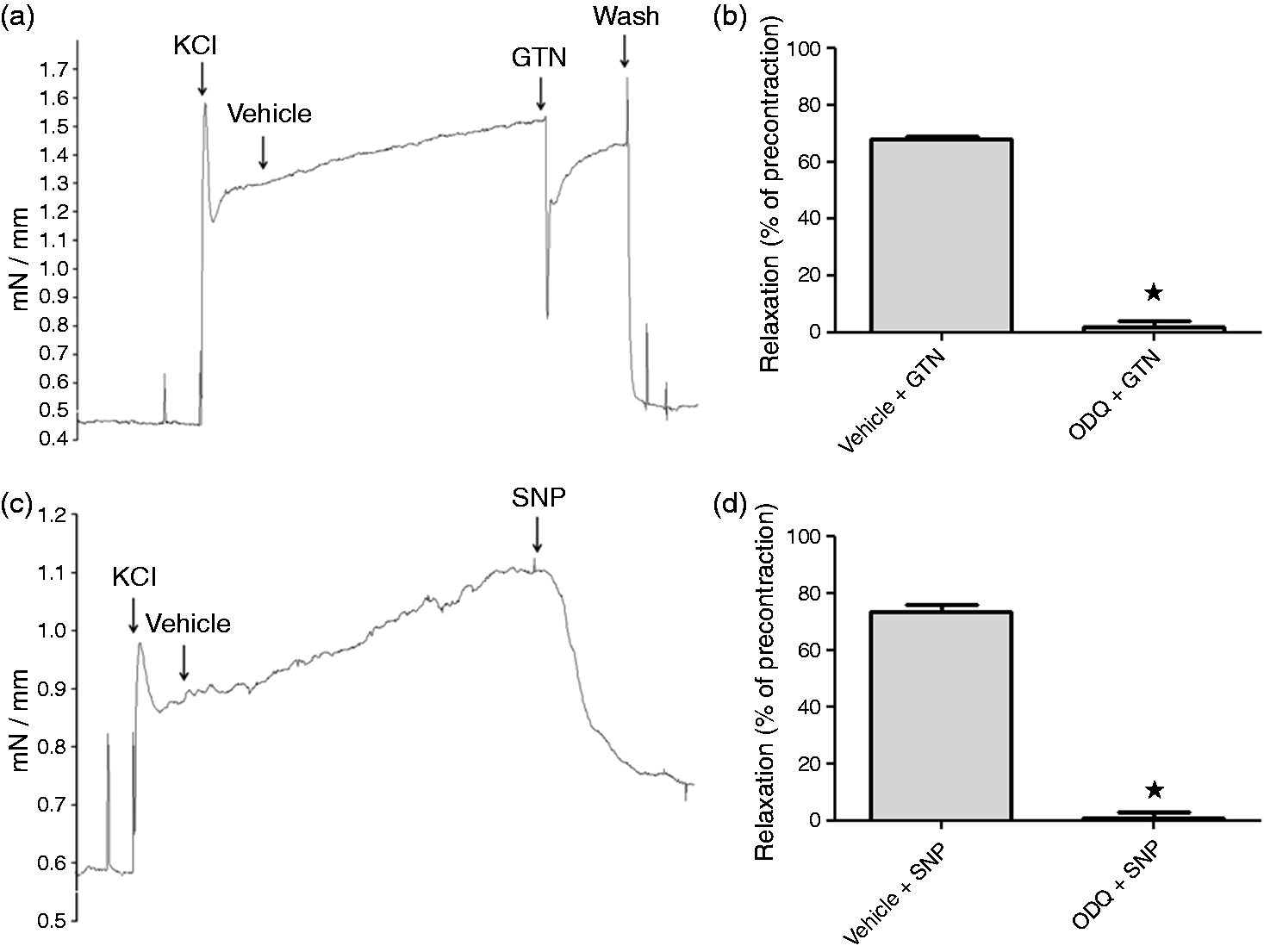
Effect of sumatriptan on GTN-induced mast cell degranulation in vivo
Pre-treatment with sumatriptan significantly inhibited GTN-induced degranulation of dural mast cells, resulting in a 21 ± 5% (n = 7) degranulation, which is statistically equivalent to the basal level of 19 ± 6% (n = 8). In comparison, GTN infusion without pre-treatment resulted in a 52 ± 8% degranulation (p = 0.0021, n = 9, Figure 7).
Effect of sumatriptan on GTN induced mast cell (MC) degranulation. GTN-induced dural MC degranulation in sumatriptan pre-treated rats. The level of degranulation is given as percentage of degranulated cells to the total number of counted cells. Vehicle pre-treated, vehicle infused animals and sumatriptan pre-treated, GTN-infused animals did not show any significant differences. # Represents significant differences from vehicle infusion whereas * represents significant difference from GTN treatment (one-way ANOVA, p = 0.0021, Bonferroni's multiple comparison test). Please note that the vehicle and GTN treated animals are the same as in Figures 4 and 9. Values are given as mean ± SEM, n = 7–9.
Effect of sumatriptan on compound 48/80 induced mast cell degranulation ex vivo
Ex vivo administration of the mast cell degranulator, compound 48/80 (1 µM), significantly increased the level of degranulated mast cells from a basal level of 11 ± 4% to 40 ± 6% after 10 min incubation (p < 0.0001; n = 8). In the presence of sumatriptan (30 µM), the level of degranulation was 8 ± 2 % (n = 5), which was similar to the level of degranulation in vehicle treated dura mater. Pre-treatment with sumatriptan did not affect the mast cell degranulation induced by compound 48/80 treatment (46 ± 8% degranulation, n = 5, Figure 8).
Ex vivo effect of sumatriptan on compound 48/80 induced mast cell (MC) degranulation. Compound 48/80 (1 µM) causes significant MC degranulation after 10 min ex vivo as compared with baseline level. Dura mater was subjected to sumatriptan (suma) for 10 min followed by addition of either vehicle or compound 48/80 for another 10 min. Suma did not cause any degranulation by itself nor prevents compound 48/80 to induce degranulation. The levels of intact and degranulated mast cells were counted under microscope and degranulation levels are given as percentage of degranulated mast cells of the total number of counted mast cells in 10 visual fields. * Indicates significant difference to vehicle; # Indicates significant difference to suma treated dura mater (p < 0.05). One-way ANOVA, p < 0.0001, followed by Dunnett’s multiple comparison test. Values are given as mean ± SEM, n = 5.
Effect of olcegepant and L-733,060 on GTN-induced mast cell degranulation in vivo
To investigate if mast cell degranulation might be due to neuronal CGRP and substance P release, we pre-treated the animals with olcegepant or L-733,060. The level of mast cell degranulation in GTN-treated rats was 52 ± 8% (n = 9). In olcegepant pre-treated animals, the GTN provoked degranulation was 60 ± 13% (n = 7) and in rats pre-treated with the NK-1 receptor antagonist L-733,060 the level of degranulation was 61 ± 10% (n = 7). Both pre-treatments were statistically equivalent (p > 0.05) to the GTN provoked group and thereby not preventing mast cell degranulation (Figure 9).
Effect of olcegepant and L-733,060 on GTN induced mast cell (MC) degranulation. GTN-induced dural MC degranulation of rats pre-treated with the CGRP receptor antagonist olcegepant or the substance P receptor antagonist L-733,060. The level of degranulation is given as percentage of degranulated cells of the total number of counted cells. Neither olcegepant nor L-733,060 pre-treated, GTN-infused groups showed any differences from vehicle pre-treated, GTN infused groups, whereas they all differed significantly from vehicle treated animals indicated by * (p < 0.05, one-way ANOVA, p = 0.0095, Dunnett’s multiple comparison test). Please note that the vehicle and GTN treated groups are the same as in Figures 4 and 7. Values are given as mean ± SEM, n = 8–9.
Discussion
In the present study we demonstrate that mast cells in the dura mater degranulate in response to GTN infusion in vivo and that this effect can be aborted by pre-treatment with the anti-migraine drug sumatriptan and by non-selective NOS inhibition, which has also proven effective in anti-migraine treatment (21). Antagonists of CGRP and substance P receptors both failed to inhibit GTN-infusion induced mast cell degranulation. Neither sumatriptan nor NO donated from GTN or SNP had a direct effect on mast cell degranulation when applied topically on the dura mater ex vivo.
Mast cell degranulation after GTN infusion
We found mast cells in the dura mater to mainly reside close to blood vessels as previously described (5). Mast cells have also been demonstrated in the proximity of primary afferent neurons (3–5), suggesting that degranulation of mast cells could initiate an inflammatory response exciting meningeal sensory afferents. GTN infusion in a physiologically relevant dose to awake rats induced a time-dependent mast cell degranulation in dura mater. We observed a non-significant increase in degranulation 30 min after GTN infusion and a prominent 6-fold increase at 2 hours that was maintained for at least 6 hours. Due to a half-life of NO in the range of minutes (22), this delayed degranulatory effect several hours after GTN stimulation indicates that mast cell degranulation is secondary to NO donated from GTN. The recovery time for mast cells is up to 48 hours (23,24) and mast cell degranulation could, theoretically, be responsible for the long-lasting neuronal activation found after GTN-infusion (9,25,26). In some of these studies, the dura mater is exposed and, consequently, mast cells are depleted and, thus, cannot influence the neuronal firing measured after GTN infusion (25,26). Previously, a neurogenic inflammation in dura mater was induced by electrical stimulation of the trigeminal nerve (27,28). This involved dilation of dural arteries, increased vascular permeability, activation of perivascular sensory nerve fibres and mast cell degranulation together leading to protein extravasation and inflammatory responses (4,6). It was later suggested that the degranulation of mast cells after administration of compound 48/80 was responsible for a prolonged state of excitation of trigeminal meningeal nociceptors (9). In line with these studies, infusion of GTN to anaesthetized rats was shown to induce meningeal inflammation involving degranulation of dural mast cells (14).
The role of nitric oxide after GTN infusion
Infusion of GTN in our rat model caused an increased level of nNOS containing nerve fibres in dura mater (17). The inhibitory effect of L-NAME on cFos up-regulation (17) and mast cell degranulation after GTN infusion suggests that exogenous NO administration commences an endogenous NO production and a loop of self-reinforcement of NO is involved up-stream to mast cell degranulation. At the L-NAME dose and time of infusion used in this study, a 48% increase in Mean Arterial Blood Pressure (MABP) was observed (17). It could be speculated that the hypertension might protect the mast cells from degranulation. However, this speculation is opposed by the finding of the hypertensive agent endothelin-1 to be a potent mast cell degranulator of cardiac mast cells (29). In order to exclude the possibility of a direct mast cell degranulating effect of NO, we treated dura mater with the NO donors GTN and SNP. We used 1 µM in our ex vivo experiments, which is close to the calculated concentration of GTN in the blood of the rat, which should be between 0.3 and 6 µM depending on the rates of degradation and elimination. The incubation time was chosen according to previous studies showing a maximum direct effect on mast cell degranulation after 10 min (7). In line with previous studies performed on dural mast cells using the NONOate, we did not find that GTN and SNP induced mast cell degranulation ex vivo (30). In order to verify that the absence of mast cell degranulation after GTN administration ex vivo was not due to a lack of enzymatic cleavage of GTN, we tested the NO donor, SNP, which releases NO independent of enzymatic bio-degradation. Also, SNP failed to display a degranulatory effect on dural mast cells. As a positive control of the NO release from these two NO donors, we examined their effect on isolated rat cerebral arteries and found a significant relaxation of the arteries mediated via cyclic guanosine monophosphate (cGMP) formation. Taken together, we believe that mast cell degranulation is secondary to NO released from GTN, but involves an endogenous production of NO. Such a response was found in rat cortex during and up to 1 hour after termination of a 20 min infusion period of a low GTN dose (31). A decrease in superoxide levels occurred concomitantly with the rise in NO levels. Interestingly, both responses were reversed in sumatriptan-treated rats. Thus, involvement of reactive oxygen species (ROS) cannot be excluded.
The role of sumatriptan on GTN-induced mast cell degranulation
The very effective and widely used anti-migraine drug sumatriptan inhibited GTN-induced mast cell degranulation. As sumatriptan probably does not pass the blood–brain barrier due to high water solubility (32), it may act by inhibiting vasodilation of meningeal (dural) arteries (19) and/or inhibition of neuropeptide release from sensory nerve endings in dura mater (33). However, another possibility could be a direct inhibition of mast cell degranulation. To the best of our knowledge, the presence of 5-HT1B and 5-HT1D receptors have not been studied on dural mast cells. 5HT1B receptors have, however, been shown in mast cells of the caput epididymis (34). We found that sumatriptan has no direct inhibitory effect on IgE-independent compound 48/80 induced mast cell degranulation ex vivo, indicating that sumatriptan has its action up-stream to mast cell degranulation. Sumatriptan has been shown to initiate a direct excitatory effect on dural afferents that led to the suggestion that it caused the initial worsening of headache observed shortly after sumatriptan treatment (35). However, as sumatriptan in our control experiments did not cause an increase in degranulated mast cells (Figure 8), we believe that this effect does not involve mast cell degranulation. Yet another possible mode of action for sumatriptan could be via scavenging of ROS as described previously (36). However, the source of dural ROS after GTN infusion is still to be investigated, as ROS probably is not released from dural mast cells, as these are intact after sumatriptan pre-treatment. Furthermore, the lack of effect of olcegepant and L-733,060 on GTN induced mast cell degranulation, as discussed below, also indicates that the effect of sumatriptan in this model is not via inhibition of CGRP and substance P release.
The role of sensory neuropeptides on GTN induced mast cell degranulation ex vivo
There is an increase in the density of CGRP containing nerve fibres in dura mater after GTN infusion as shown in Figure 1 and by Ramachandran and colleagues (17). At the time of designing this study, discrepancy existed between studies showing the CGRP releasing effect of NO-donors. In one ex vivo study, the NO donor NONOate releases CGRP from dura mater in a rat skull model (18). Thus, it can be speculated that NO could promote an increased release of CGRP and substance P from sensory nerve terminals leading to mast cell degranulation via activation of CGRP- and NK1 receptors (6,37). In another study performed on skulls from guinea pigs GTN and SNP failed to release CGRP (38). However, in a recent paper, published after the performance of these experiments, NO was shown to react with H2S, leading to the generation HNO, which activates the TRPA1 receptor to release CGRP from dural sensory neurons (39). As NK1 receptor- and CGRP receptor antagonists have previously been shown to inhibit substance P and CGRP induced mast cell degranulation in rat (6), we aimed to investigate if GTN infusion causes receptor-mediated mast cell degranulation via release of CGRP and substance P. Rats were pre-treated with the CGRP antagonist olcegepant and the NK1 receptor antagonist L-733,060, both of which have been shown to inhibit GTN induced increase in trigeminus nucleus caudalis c-fos expression (17). The two antagonists had no effect on GTN induced mast cell degranulation in vivo although the doses used have an inhibitory effect on GTN induced c-fos expression (17). This finding further supports our previous studies showing that there is no increase in CGRP release after GTN infusion (40,41) and that NO is not causing release of CGRP from dural sensory nerve terminals (38). On the other hand, the inflammatory mediators released during GTN induced mast cell degranulation may be responsible for a peripheral sensitization due to p-ERK up-regulation as shown in rat dural sensory nerve fibres (9) and the delayed migraine attack found in migraineurs after GTN infusion (10).
Conclusion
GTN induced mast cell degranulation is not mediated via peripheral activation of sensory neurons. Hence, CGRP and substance P seems to have a role in GTN infusion induced c-fos activation via another mechanism. NOS activation and 5-HT1B/1D receptors are involved in GTN-infusion induced mast cell degranulation in vivo via yet unknown mechanisms.
Article highlights
In vivo administration of glyceryl trinitrate (GTN) to freely moving rats causes dural mast cell degranulation This effect involves endogenous nitric oxide production and can be inhibited by pre-treatment with non-selective nitric oxide synthase inhibition (L-NAME) and sumatriptan GTN induced degranulation is not mediated via CGRP release from sensory neurons
Footnotes
Funding
This work received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
Jes Olesen has within the last 2 years received grants/research support from and/or has been a consultant/scientific advisor for, and/or has been on the speaker’s bureau of Alder Biopharma, Proreo Pharma and Amgen.
Acknowledgements
This study was supported by the The Lundbeck Foundation (R77-A6952), Candy’s Foundation, The Danish Research Council (11-107831) and the Novo Nordisk Foundation, Frimodt-Heineke Foundation, Else and Mogens Wedell-Wedellsborgs Foundation, The A.P. Møller Foundation for the Advancement of Medical Science. The authors would like to thank Elisabeth Sahlén for valuable laboratory assistance.