
Abstract
Background
Obesity is a risk factor for episodic migraine to develop into chronic migraine; hence, it is speculated that obesity and hyperleptinemia are associated with migraine. We hypothesized that leptin is involved in the mechanisms of cortical spreading depression (CSD). Therefore, we examined whether leptin affected a rat model of CSD to clarify the relationship between leptin and migraine.
Methods
We evaluated the effect of intracerebroventricular (ICV) administration of leptin on a rat CSD model. We then examined whether once-a-day intraperitoneal administration of leptin for seven days (as a chronic hyperleptinemia model) affected rat CSD models. Finally, we induced CSD in Zucker fatty (ZF) rats, which is a well-known model of obesity.
Results
In the parietal cortex, the percent change in cerebral blood flow and direct current (DC) potential decreased after ICV administration of leptin. A similar decrease in DC potential was observed in rats treated with intraperitoneal leptin. The number of CSDs increased significantly in rats given intraperitoneal leptin and in ZF rats.
Conclusions
The present study suggests that leptin is involved in the mechanisms of CSD.
Introduction
Previous reports regarding the pathogenesis of migraine focus on body weight and abdominal obesity (1). Although migraine prevalence does not vary with body mass index, the risk of chronic migraine is higher in obese migraineurs experiencing episodic migraines (2). Individuals with episodic migraine who are obese have more severe and debilitating migraine attacks and more often experience photophobia and phonophobia (3). In particular, morbidly obese women show a higher incidence of migraine with aura (4). It is speculated that several adipocytokines, such as leptin, play an integral role in feeding, and subsequently obesity, and that obesity is associated with migraine (5).
Leptin is a product of the obese (ob) gene and consists of 167 amino acid peptides. Leptin is primarily produced by adipocytes, and is produced in several tissues including the stomach, muscle, bone marrow, and the brain (5). Leptin receptors are abundantly expressed in the arcuate nucleus, lateral area, paraventricular nucleus, dorsomedial nucleus, and ventromedial nucleus of the hypothalamus (6). Leptin secreted by adipose cells is stimulated by feeding and insulin administration, and its secretion volume has been correlated with body fat (5). Mice carrying mutations in the ob gene or the gene encoding the leptin receptor (i.e. db/db) express an obese phenotype (7). Leptin induces production of nitric oxide, several cytokines (e.g. tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6)), and calcitonin gene-related peptide (CGRP) (8,9), and each of these substances has been associated with the pathological mechanisms of migraine (10). Furthermore, leptin is involved in the pathogenesis of neuropathic pain in rodents (11).
Cortical spreading depression (CSD) is a slowly spreading wave of neuronal and glial depolarization that is triggered by electrical or chemical stimulation. In animal experiments, cerebral blood flow (CBF) increases transiently with CSD, but decreases afterward for several hours (12–14). CSD is considered a mechanism of aura and may help initiate a migraine attack (15). CSD has been identified in the human visual cortex with functional magnetic resonance imaging (16). However, the effect of leptin on CSD has not been investigated.
We therefore hypothesized that leptin modulates the pathological mechanisms of migraine and that chronic hyperleptinemia changes cortical excitability and susceptibility to CSD. Leptin induces the production of several cytokines, and an increase in inflammatory cytokines (e.g. TNF-α or IL-6) could cause epilepsy (17), which is related to neuronal hyperexcitability. To evaluate our hypotheses, we developed three experimental protocols that examined whether hyperleptinemia significantly increased the occurrence of CSD. First, we examined whether intracerebroventricular (ICV) leptin administration affected a rat CSD model. We then examined whether chronic hyperleptinemia, which was induced with once-a-day intraperitoneal leptin administration for seven days, affected a rat CSD model. Together, these two experiments examine whether CSD is induced by extrinsic leptin. Finally, we induced CSD in the Zucker fatty (ZF) rat (18), which is a model of human obesity that shows intrinsic hyperleptinemia, and then compared our findings with those from the two previous experiments to clarify the relationships among obese migraineurs’ CSD and leptin.
Methods
Animals
The study protocol was approved by the Animal Ethics Review Committee of Kitasato University, and was conducted in compliance with the Guidelines for Proper Conduct of Animal Experiments (2006) approved by the Science Council of Japan. Sprague Dawley rats (Oriental Yeast Co., Tokyo, Japan), ZF rats and Zucker lean (ZL) rats (Charles River Japan Inc, Yokohama, Japan) were used in this experiment. Rats were kept under standard laboratory conditions with tap water and regular rat chow available ad libitium; lights were maintained on a 12-hour light-dark cycle.
CSD induction
In each rat, a tracheostomy was performed with controlled ventilation (Shinano Mfg., Tokyo, Japan) and under intraperitoneal anesthesia induced with a mixture of α-chloralose (60 mg/kg; Wako, Osaka, Japan) and urethane (600 mg/kg; Showa Chemical Industry, Tokyo, Japan) in saline. After catheterizing the right femoral artery, the heart rate and mean arterial blood pressure were measured via a pressure transducer (NEC San-ei Instruments, Tokyo, Japan). A laser-Doppler probe was placed on the cerebral cortex through a left bone fenestration located 2 mm posterior and 2 mm left to bregma to measure CBF (ALF-21N; Advance Inc, Tokyo, Japan). A platinum electrode coated with chloroplatinic acid was used to measure the direct current (DC) potential, which was inserted through a left bone fenestration located 2 mm posterior and 4 mm lateral (left) to bregma. The Ag/AgCl reference electrode was inserted into the temporal muscle. The potential between a platinum electrode and an Ag/AgCl electrode placed in the temporal muscle was recorded during the experiment. One bone fenestration was opened at a site 7 mm posterior and 3 mm lateral (left) to bregma to apply KCl solution. We maintained the mean arterial blood pressure within the range of 110 ± 5 mmHg. Rectal temperature was maintained at approximately 37℃ by using a warmer (A.S. One Co., Osaka, Japan). PaCO2 was maintained within the range of 40 ± 1.0 Torr, which was measured from blood samples obtained from the femoral artery (ABL 505; Radiometer, Copenhagen, Denmark). Thirty minutes of a stable potential was allowed after DC potential electrode insertion, and then 1.0 M KCl solution was applied through the bone fenestration onto the cortical surface to induce CSD (14). CBF amplitude was evaluated after KCl application; variations in CBF were quantified by the percent change in CBF. A change in DC potential from base to peak was also evaluated after KCl application; variations in DC potential were quantified by the percent change in DC potential. The number and duration of CSDs also were recorded. The total CSD duration was determined by summing the durations of individual CSDs.
Rat CSD model for ICV administration of leptin
Seven male Sprague Dawley rats weighing 452.5 ± 5.7 g were used in this experiment. In each rat, a tracheostomy with controlled ventilation and intraperitoneal anesthesia was performed using the same methods described above. A 30-gauge stainless steel cannula was positioned stereotaxically in the right hemisphere (3.6 mm vertically from the brain surface) for ICV administration of leptin. The cannula was inserted through a burr hole located 1 mm posterior and 2 mm lateral to bregma. In each experimental session, 20 mM Tris buffer (pH 7.5) was given via ICV administration, and then 1.0 M KCl solution was applied through the bone fenestration on the cortical surface to induce CSD (Figure 1(a)). After this application, we observed CSD for one hour. Following the disappearance of CSD, 1.0 M KCl solution was again applied through the bone fenestration following ICV administration of leptin (6 µg in 5 μl; Sigma-Aldrich, St Louis, MO, USA) to induce CSD (Figure 1(b)) (19). The percent change in CBF, the percent change in DC potential, number of CSDs, and duration of CSD were recorded.
Rat CSD model with intracerebroventricular administration of leptin. Comparisons of CSD before (a) and after (b) leptin administration. The mean percent change in CBF (a) and DC potential (b) significantly decreased after leptin treatment. The number of CSDs (c) and the duration of CSD (d) showed no significant difference between groups. Data are shown as the mean ± SEM. *p < 0.05, paired t-test, n = 7. CSD: cortical spreading depression; CBF: cerebral blood flow; DC: direct current.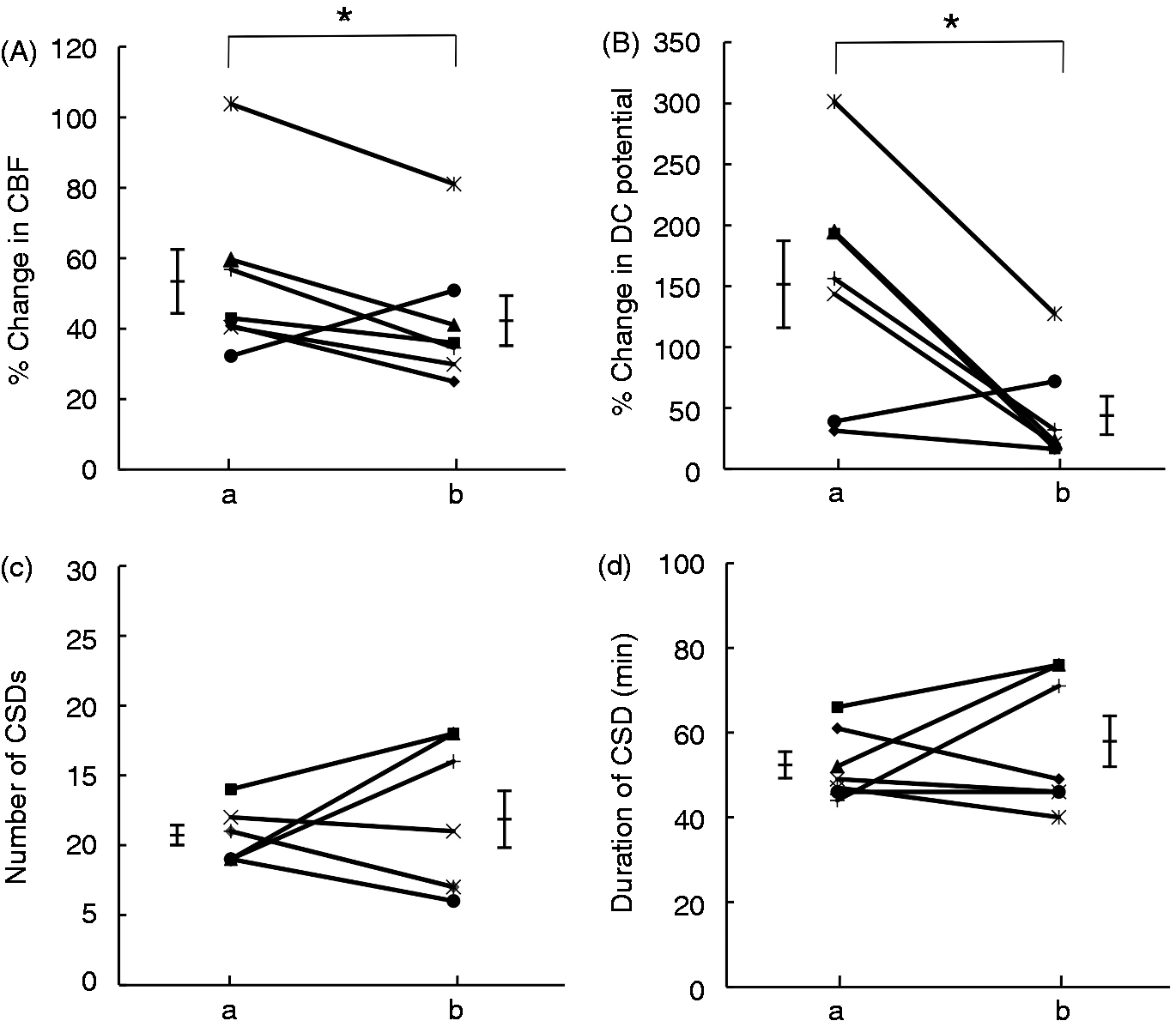
Rat CSD model with intraperitoneal administration of leptin
Fourteen male Sprague Dawley rats were used in this experiment. Leptin (0.1 mg·kg−1·day−1) or saline was administered intraperitoneally once a day for seven days (n = 7) (20), and CSD was induced on day eight. The mean body weight was 432.5 ± 8.23 g for rats administered leptin and 439.8 ± 8.6 g for rats administered saline (weighed on day eight). The percent change in CBF, the percent change in DC potential, number of CSDs, and duration of CSD were measured.
CSD model of ZF rat
Six male ZF rats (455 ± 9.2 g) were used as animal models of obesity and chronic hyperleptinemia; six male ZL rats (335 ± 17.1 g) were used as controls. We induced CSD both in ZF and ZL rats using our methods described above and then measured the percent change in CBF, the percent change in DC potential, number of CSDs, and duration of CSD.
Statistical analysis
Values for group means are presented as mean ± SEM. The statistical significance of comparisons between groups was determined with paired t-tests (StatMate; ATMS Co., Tokyo, Japan) for the ICV leptin-treated rats and unpaired t-tests for the intraperitoneal leptin-treated rats and ZF rats. A p value < 0.05 was considered statistically significant for all comparisons.
Results
CSD following ICV administration of leptin
Following ICV administration of leptin, the mean percent change in CBF (42.6 ± 7.1%) decreased significantly from pretreatment levels (53.8 ± 9.1%, p = 0.042). The percent change in DC potential (44.0 ± 15.7%) was also significantly less than that measured before leptin treatment (151.4 ± 35.6%, p = 0.015). However, the number of CSDs did not change with ICV administration (10.7 ± 0.7), as compared with pretreatment levels (11.9 ± 2.0, p = 0.599). Moreover, the duration of CSD did not change with ICV administration (52.1 ± 3.1 min), as compared with pretreatment levels (57.7 ± 6.0 min, p = 0.370; Figure 1).
CSD following intraperitoneal administration of leptin
We observed no significant difference in the mean percent change in CBF between intraperitoneal leptin administration (54.5 ± 5.5%) and vehicle treatment (71.4 ± 12.2%, p = 0.232). The mean percent change in DC potential significantly decreased with leptin administration (15.5 ± 2.9%) as compared with vehicle treatment (28.3 ± 5.3%, p = 0.027). The mean number of CSDs was significantly increased with leptin administration (14.1 ± 1.0), as compared with vehicle treatment (9.9 ± 0.7, p = 0.004). In contrast, there was no significant difference in the mean duration of CSD between intraperitoneal leptin (49.9 ± 4.0 min) and vehicle administration (46.1 ± 3.9 min, p = 0.520; Figure 2). The body weights of rats receiving leptin and their amount of food intake remained unchanged after one week.
Rat CSD model with intraperitoneal leptin administration. The mean percent change in CBF (a) shows a tendency to decrease when compared with vehicle treatment. The mean percent change in DC (b) potential was significantly less than that measured in the vehicle group. The number of CSDs (c) was significantly greater than that in vehicle rats. However, the duration of CSD (d) was not significantly different. Data are shown as the mean ± SEM. *p < 0.05, **p < 0.01, unpaired t-test, n = 7. CSD: cortical spreading depression; CBF: cerebral blood flow; DC: direct current.
Investigation of ZF rat CSD
We observed no significant difference in the mean percent change in CBF between ZF (53.4 ± 7.8%) and ZL (73.0 ± 11.3%, p = 0.185) rats. The mean percent change in DC potential significantly increased in ZF rats (29.3 ± 5.1%) as compared to ZL rats (15.8 ± 2.5%, p = 0.039). The mean number of CSDs (25.3 ± 3.4) in ZF rats was significantly greater than those in ZL rats (10.2 ± 1.2, p = 0.002). The mean duration of CSD in ZF rats (83.5 ± 11.3 min) was significantly longer than those in ZL rats (43.3 ± 4.5 min, p = 0.008; Figures 3 and 4).
The number and duration of CSD events. The number and duration of CSD events were significantly increased in Zucker fatty (ZF) rats, as compared with Zucker lean (ZL) rats (control). (a) CBF in ZL rat, (b) DC potential in ZL rat, (c) CBF in ZF rat, (d) DC potential in ZF rat. CSD: cortical spreading depression; CBF: cerebral blood flow; DC: direct current. CSD model in ZF rats. No significant difference was observed between ZF rats and ZL rats for the mean percent change in CBF (a). The mean percent change in DC potentials (b), the number of CSDs (c), and the duration of CSD (d) were significantly increased in ZF rats as compared with ZL rats. Data are shown as the mean ± SEM. *p < 0.05, **p < 0.01, unpaired t-test, n = 6. CSD: cortical spreading depression; CBF: cerebral blood flow; DC: direct current; ZF: Zucker fatty; ZL: Zucker lean.

Discussion
In the present study, we examined the effect of leptin on a rat CSD model to clarify the relationship between leptin and CSD. The DC potential was significantly decreased following both ICV and intraperitoneal leptin administration, but it was significantly increased in ZF rats. The number of CSDs increased significantly in rats receiving intraperitoneal leptin and ZF rats. Based on these findings, the susceptibility of the cortex to produce CSD may increase in chronic hyperleptinemia.
Leptin, the product of the ob gene, has been suggested to reflect fat mass (5). The leptin receptor, Ob-R, is a member of the class 1 cytokine receptor family, is expressed in 6 isoforms (Ob-Ra–Ob-Rf), and is produced by alternative RNA splicing of the db gene. Ob-Rb mainly transcribes activation signals within cells (21). Furthermore, leptin administration in rats and mice reduces feeding and body weight, but increases energy expenditure (22,23). In our experiments, however, rats receiving intraperitoneal leptin administration for one week did not show a significant body weight reduction. It is possible that their body weights might have been reduced after long-term leptin administration that exceeded one week.
The mechanisms underlying CSD-evoked changes in CBF and DC potential are complex and involve several chemical factors. CSD is a transient depolarization of nerve cells. In the brain, depolarization occurs with varying DC potentials in response to damage or physicochemical stimulations (e.g. KCl injection). From the focus of the local depolarization, a wave of depolarization spreads slowly at a rate of 2–5 mm/minute and repeats (12,13). In ZF rats, depolarization that is induced by KCl may be enhanced, and then the mean percent change in DC potential also may be increased. In contrast to this result, the mean percent change in CBF and DC potential were decreased after ICV administration of leptin. Depolarization may be unlikely to occur following a single administration of leptin. As a result, the number of CSDs might not change after ICV administration of leptin. There is a possibility that a single leptin injection may suppress the induction of CSD compared with chronic hyperleptinemia. Compared with ZF rats, the mean percent change in DC potential was decreased after intraperitoneal leptin administration, but the number of CSDs was increased, the same as ZF rats.
Our results suggest that the number of CSDs may be increased in chronic hyperleptinemia. Nevertheless, the relationship between the number of CSDs and the severity of migraine is uncertain. Factors or conditions that increase neuronal excitability appear to increase the frequency of CSDs (24). Leptin can directly inhibit serotonin production and release from the raphe nuclei of the brainstem (25). The frequency of CSD waves increases with lower serotonin levels (24). We have previously shown that orexin-A inhibits the occurrence of CSD (26–28), and it is known that leptin can inhibit orexin-A secretion (29). The serotonergic system and secretion of orexin-A could be inhibited by hyperleptinemia, and as a result, CSD may be enhanced.
Changes in CSD frequency may reflect changing excitability in the cerebral cortex and the susceptibility to migraine attacks. A facilitating effect of CSD on synaptic excitability may contribute to neocortical hyperexcitability (12). The induction of CSD significantly increased the amplitude of field excitatory post-synaptic potentials and increased long-term potentiation (LTP) in the third layer of human neocortex (30). Leptin facilitated LTP in the hippocampal CA1 field of rats with intact signaling and enhanced emotional and spatial learning (31,32). However, there are some reports that leptin receptor-deficient animals show impaired LTP in CA1 that is attributed, at least in part, to a deficiency in leptin receptors in the hippocampus (33,34). CSD occurs in the cortex as well as in many other regions of the brain. The leptin receptor is also expressed in the cortex as well as in many other regions of the brain. Therefore, it is difficult to explain the cause of cortical hyperexcitability by only an LTP-like phenomenon. An increase in inflammatory cytokines (e.g. TNF-α or IL-6) can cause epilepsy (17), and therefore, cortical hyperexcitability might be increased by these inflammatory cytokines, which are elevated in ZF rats.
Exactly how leptin changes the susceptibility to CSD is unknown. Astrocytes continue to regulate potassium and calcium during CSD (35). Thus, clearance of KCl, which can elicit CSD, depends mainly on astrocytes. Astrocytic Ob-R expression is present in rodents, and astrocytic Ob-R is related to the neuroinflammatory response (36). Furthermore, astrocytic Ob-R protein was identified in the rat hypothalamus using double-labeling immunohistochemistry (36). In hypothalamic neurons, such as those in the arcuate and ventromedial nuclei, KATP channels are activated by Ob-R (37). The brain regions that express astrocytic Ob-R, however, are still unknown. The hypothalamus is involved in the pathogenesis of migraine; therefore, astrocytic Ob-R in the hypothalamus may have some relationship with migraine. Astrocyte activity is altered by hyperleptinemia, which may subsequently change the susceptibility to CSD.
We used ZF rats in the present study because they show hyperleptinemia that is similar to that observed in obese humans. The ZF rat has been widely used to investigate pathogenic conditions such as maladaptive feeding behaviors, obesity (hyperleptinemia), diabetes and hypertension. The number of CSDs increased significantly in rats that received an intraperitoneal administration of leptin, especially the ZF rats. Compared with ZL rats, ZF rats have leptin receptor abnormalities, a large fat mass and an abundance of pro-inflammatory cytokines. Leptin induces nitric oxide production, cytokines (e.g. TNF-α or IL-6) and CGRP; each of these substances could increase the number of CSDs. ZF (fa/fa) rats carry an autosomal-recessive mutation in the gene that encodes the leptin receptor, and as a result, present with hyperleptinemia (18). The fa (leptin receptor) mutation (Gln269Pro) affects the extracellular domain common to all isoforms, but its impact on leptin activity is uncertain (38). The leptin-mediated effects on neuropeptide expression, signal transduction and feeding behaviors are reduced but not abolished in ZF rats (39,40). Leptin receptors other than Ob-Rb also may be related to CSD. Therefore, it is possible that the number of CSDs occurring in ZF rats is increased. ZF rats show abnormal carbohydrate and lipid metabolism (2,5), and this abnormal metabolism may also affect CSD. The reason why the number of CSDs did not change after ICV leptin administration may be that a single administration of leptin is insufficient for changing neuron and astrocyte excitability. These results suggest that chronic hyperleptinemia may increase the frequency of CSDs.
Conclusion
The present study suggests that leptin is involved in the mechanisms of CSD. Chronic hyperleptinemia may cause a change in cortical excitability and the susceptibility to CSD. Thus, our results suggest that episodic migraine may develop into chronic migraine through chronic hyperleptinemia.
Clinical implications
Remarkable findings of our study include:
Intracerebroventricular (ICV) administration of leptin significantly decreased percent change in cerebral blood flow compared with vehicle rats. Zucker fatty rats that express endogenously chronic hyperleptinemia show the number of CSDs and duration of CSDs increased significantly. Obesity is known as one of the risks for episodic migraine to change into chronic migraine. It is speculated that the obese person’s hyperleptinemia has some kind of association with migraine. We made a hypothesis that hyperleptinemia induces chronic migraine. Our findings suggest that leptin modulates the pathological mechanisms of CSD.
Footnotes
Funding
This research received no funding from any agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.
Ethics or institutional review board approval
The study protocol was approved by the Animal Ethics Review Committee of Kitasato University, and was conducted in compliance with the Guidelines for Proper Conduct of Animal Experiments (2006) approved by the Science Council of Japan.