
Abstract
Objective: To evaluate the chronic effect of topiramate (TPM) on cortical spreading depression (CSD), which is thought to be related to migraine aura.
Methods: Male rats (n = 30) were randomized to once-daily peroral treatment with TPM (50, 100, 200 or 600 mg/kg) or vehicle for 6 weeks. We evaluated the characteristics of CSD induced by topical application of KCl under isoflurane anesthesia and the changes in plasma level of TPM in each group. The effect of single administration of TPM on CSD was also evaluated.
Results: After the final administration of TPM, when the plasma level of TPM was high, KCl-induced CSD frequency and CSD propagation velocity were dose-dependently reduced and the interval between CSD episodes was elongated, compared with the vehicle control. However, before the final administration of TPM, when the plasma level was very low, the KCl-induced CSD profile was the same as that in the vehicle control. Single administration of TPM did not alter the CSD profile. Local cerebral blood flow was not significantly altered by chronic administration of TPM.
Conclusion: TPM suppressed the frequency and propagation of CSD along the cerebral cortex, and might be a candidate for relief of migraine.
Introduction
Migraine is a common, chronic, incapacitating neurovascular disorder characterized by severe headache, autonomic nervous system dysfunction, and in some patients an aura involving neurologic symptoms (1). Recently developed selective serotonin receptor agonists, such as triptans, are useful to treat migraine, but strict compliance with the dosing schedule is essential for effective treatment. Furthermore, frequent migraine attacks may lead to excessive acute medication without proper supervision, potentially resulting in medication-overuse headache (2). Therefore, prophylaxis of migraine is an important goal.
The pathophysiology of migraine is very complex, but one of the main correlates is thought to be cortical spreading depression (CSD) (3,4). CSD is a transient neuronal depolarization that slowly propagates along the cerebral cortex, followed by long-lasting suppression of neuronal activity (5). It has been proposed that CSD is a neuronal mechanism underlying migraine aura (6) and is involved in vasodilation of the middle meningeal artery during headache, which is linked to changes of neurometabolic brain activity with transmission via the trigeminal nerve (7).
A β-adrenoceptor antagonist, propranolol, has been widely used in the prophylaxis of human migraine (8) and has been shown to suppress CSD in an animal model (9). However, the mechanism of its action remains unclear. Topiramate (TPM; 2,3:4,5-bis-O-(1-methylethylidene)-beta-
Materials and methods
General procedures of chronic study
Animals were used with the approval (No. 09058) of the Animal Ethics Committee of Keio University (Tokyo, Japan), and all experimental procedures were in accordance with the university’s guidelines for the care and use of laboratory animals. Male Sprague-Dawley rats (8–9 weeks, initial body weight; 412 ± 28 g, n = 30) were randomized to five groups so that the mean body weights were not significantly different among groups: a vehicle group (0.5% methylcellulose: 400 cps (centipoises); Sigma Aldrich Japan, Tokyo, Japan) and four TPM groups (50 mg/kg, 100 mg/kg, 200 mg/kg, 600 mg/kg; provided by Janssen Pharmaceutical K.K., Tokyo, Japan). Test solution was adjusted to 5 ml/kg and administered by gavage once daily for 6 weeks. During the administration period, rats received food and water ad libitum. The animals were kept in an air-conditioned room maintained at 23.0 ± 1.0°C and 55 ± 7% humidity with automatic lighting between 08:00 and 20:00. Body weight was monitored daily throughout the treatment period.
Procedures of CSD measurement
On the final treatment day, the animals were anesthetized with isoflurane (2.5–3.0%) and subjected to CSD evaluation as previously reported (21). Briefly, each rat was fixed to a head-holder (SG-3 N modified to be flexible around the horizontal axis, Narishige Scientific Instrument Lab., Tokyo, Japan) and a cranial window of approximately 4 mm in diameter was opened in the left side of the skull at the parieto-temporal region of the cerebral cortex. The dura was removed and the exposed cortex was covered with a cover slip to prevent it from drying out. CSD was induced by introducing a drop (5 µl) of 1.0 M KCl solution into an additional posterior hole of 2 mm in diameter, centered at the coordinates of 7 mm posterior and 2 mm lateral to the bregma. Approximately 1 h after the first KCl application, when no further CSD had occurred for more than 10 min, vehicle or TPM was administered through an intragastric catheter. The second application of KCl was performed 1 h after the final administration of vehicle or TPM, as shown in Figure 1. Arterial blood pressure (ABP) was continuously recorded through a femoral arterial catheter via a surgical strain gage (MLT0670 and ML117, ADInstruments Pty. Ltd., Bella Vista, NSW, Australia). Heart rate (HR) was determined from the ABP wave. Continuous recordings of ABP, HR, cerebral blood flow (CBF) and direct current (DC) potential (see below) were acquired with a multi-channel recorder (PowerLab 8/30, ADInstruments Pty. Ltd.) and recorded with proprietary software (LabChart, ADInstruments Pty. Ltd.) for offline analysis. All procedures were performed at constant body temperature, maintained with a heating-pad and thermocontroller (BWT-100, Bioresearch Center Co., Ltd., Nagoya, Japan). CSD was elicited with KCl solution after confirmation that all parameters had remained stable for at least 10 min.
Plasma concentrations of TPM before and after the final administration of TPM on the day of CSD evaluation in chronically treated rats. An outlined arrow indicates the time of final intragastric administration of vehicle/TPM (t = 0) and solid arrows indicate the times of KCl application on the surface of the brain [KCl (before) at t = −1 and KCl (after) at t = 1].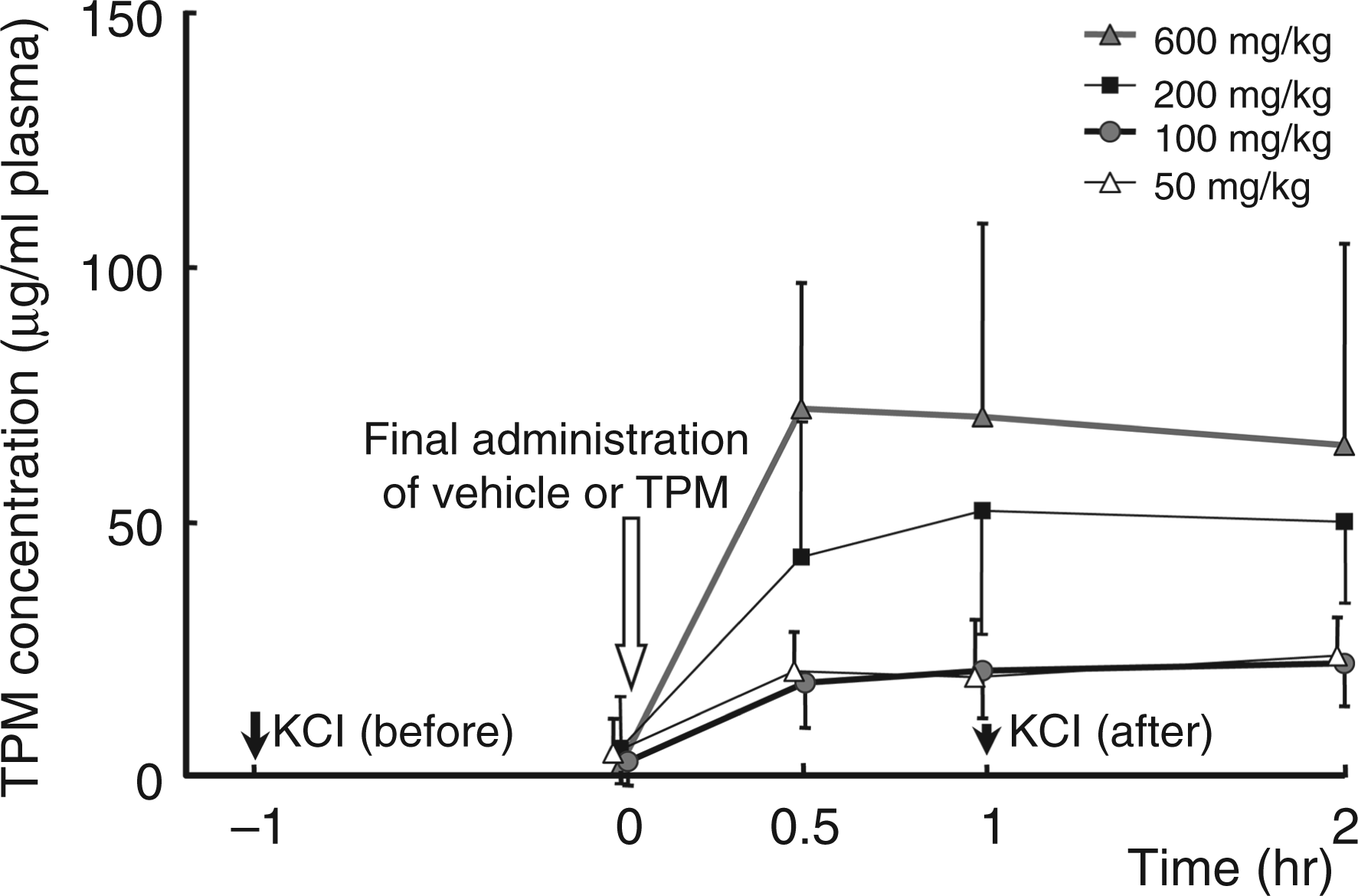
Measurements of DC potential and CBF
Two Ag/AgCl electrodes (tip diameter = 200 µm, EEG-5002Ag, Bioresearch Center Co., Ltd.) were inserted 200 µm under the pia mater at the anterior edge (0.5 mm posterior and 2 mm lateral to bregma) and posterior edge (3.5 mm posterior and 2 mm lateral to the bregma) of the cranial window, and immobilized with dental cement. Ag/AgCl reference electrodes (EER-5004Ag, Bioresearch Center Co., Ltd.) were subcutaneously placed in the space between the skull-bone and the scalp. DC potential was amplified at 1–100 Hz and digitized at 1 kHz with a differential headstage and differential extracellular amplifier (Model 4002 and EX1, Dagan Co., Minneapolis, MN, USA).
CBF was monitored with a laser Doppler flowmeter (LDF) (ALF 21 R, Advance Co., Ltd., Tokyo, Japan). The probe, which had a diameter of 0.8 mm, was placed on the surface of the cortex 2 mm posterior and 2 mm lateral to the bregma (intermediate region of the cranial window).
Evaluation of CSD susceptibility
One application of KCl induced repetitive negative deflections of DC potential (i.e., CSD). CSD was detected at the posterior site (proximal) and then at the anterior site (distal) after a delay. At the same time, CBF was elevated, as reported previously (21). We defined CSD as the occurrence of DC potential deflections detected at both sites, together with CBF elevation. We evaluated CSD occurrence frequency, interval between CSD episodes, and total length of time for which CSD continued (duration from the first CSD to the last CSD). Propagation velocity from the proximal to the distal site was also calculated based on the distance between the electrodes.
Measurement of plasma level of TPM
Before the final administration of TPM (time 0), and at 30 min, 60 min, and 120 min after the final intragastric administration of TPM, as shown in Figure 1, arterial blood was collected through the arterial catheter. After protein precipitation with acetonitrile, plasma samples were used for measurement of the plasma level of TPM by means of a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method (22) with some modifications. Separation was done on a reversed-phase LC column (X-Bridge C18 3.5 µm – 50 × 4.6 mm, Waters, Milford, USA) with a mixture of 10 mM ammonium acetate and acetonitrile as the mobile phase. Detection was by tandem MS on an API-3000 MS/MS (Applied Biosystems, Toronto, Canada), operated in the negative ion mode using the TurboIonSprayTM-interface. TPM was monitored at the m/z transition 338.1 → 78.0 using a dwell time of 300 ms. The limit of quantification was 50 ng/ml. In measurement of independent QC samples, the intra-batch accuracy was between 85% and 115%.
Acute effect of TPM on CSD
Male Sprague-Dawley rats (12–19 weeks, body weight; 519 ± 95 g, n = 7) were used for evaluation of the effect of single administration of TPM. Using the same protocol as described for chronically treated rats, we evaluated the KCl-induced CSD profile and CBF before and after intragastric administration of TPM (600 mg/kg). Changes of ABP, HR and blood gas level were followed. Arterial blood gas analysis was performed with a RapidLab 348 (Siemens AG, Munich, Germany).
Statistical analysis
All data are reported as means ± SD. Multiple comparisons were performed using Dunnett’s test among vehicle and TPM chronic administration groups. The paired t-test was used to evaluate the acute effect of TPM on the CSD profile versus the pre-administration level. A p value of <0.05 versus the vehicle treatment group or the pre-administration level was considered to be statistically significant.
Results
Plasma level of TPM
The plasma level of TPM increased rapidly after administration, and a dose-dependent plateau level was maintained for 2 h (Figure 1). After 6 weeks of daily administration, the plasma level of TPM before the final administration was 1/5 to 1/50 of the maximum level; that is, there was no indication of TPM accumulation. After intraperitoneal administration of 200 or 500 mg/kg TPM, the plasma level reached a maximum of 125 ± 14 or 155 ± 98 µg/ml, respectively, within 1 h after administration. The maximum plasma level after intragastric administration was approximately 1/3 of that after intraperitoneal administration and remained at a plateau level for a longer period.
Effect of chronic TPM on CSD
Physiological parameters just before CSD evaluation in chronically treated rats

HR, heart rate; MABP, mean arterial blood pressure.
p<0.05 significant difference from the vehicle control.
As shown in Figure 2A, one application of KCl induced repetitive (between 2 and 8 times) negative deflections of DC potential (i.e., CSD). CSD was detected first at the posterior (proximal) site, and then at the anterior (distal) site with a delay time of 14 to 80 s. After the final administration of TPM, CSD occurrence was slightly suppressed (Figure 2B). No CSD-associated change of ABP was seen in any rat. Mean arterial blood pressure (MABP) was maintained within ± 20 mmHg in each rat throughout the experiments, and did not decrease below 60 mmHg in any rat.
Original recordings of DC potential at the proximal and distal sites, as well as CBF and ABP. KCl was applied on the brain surface at the arrows, 1 h after the final administration of vehicle (A) or 600 mg/kg of TPM (B) in rats treated for 6 weeks.
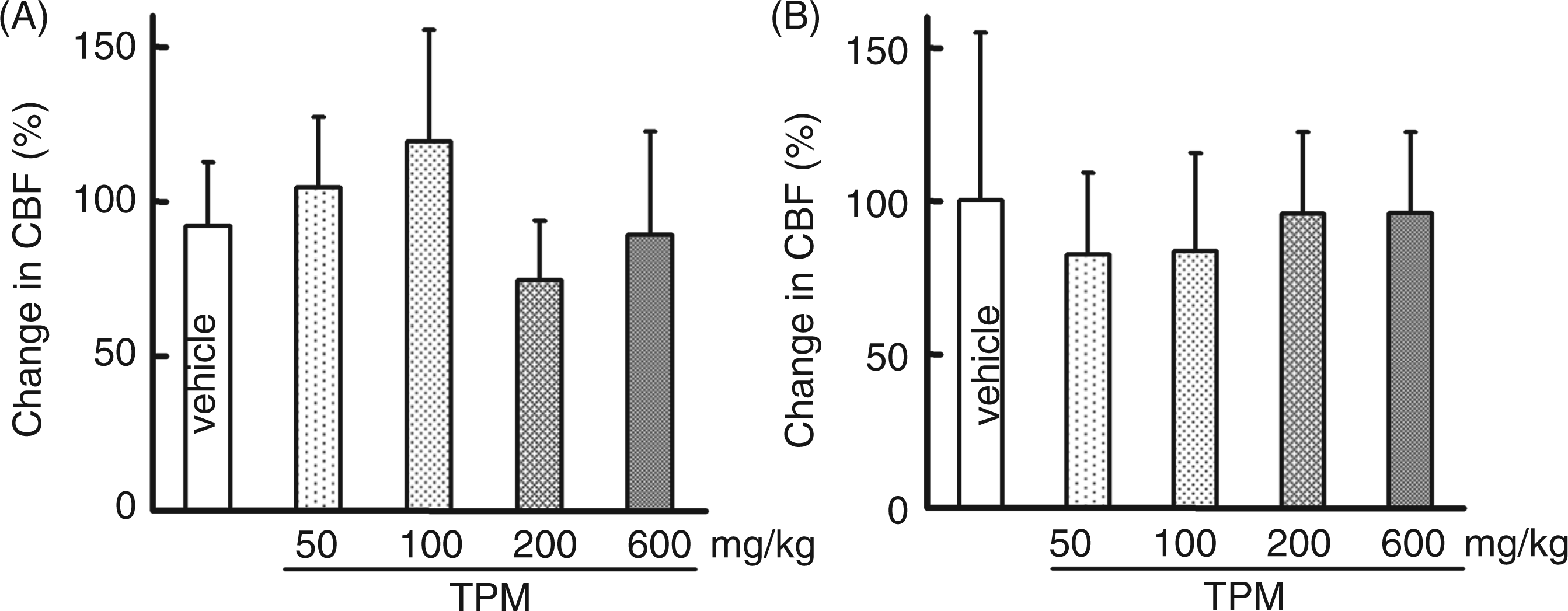
In the observation 1 h before the final oral administration of TPM, none of CSD occurrence frequency, inter-CSD interval, total length of time for which CSD continued or propagation velocity was statistically significantly different from the value in the vehicle group (Figure 3). However, 1 h after the final oral administration of TPM, during the plasma TPM level plateau period, CSD occurrence frequency was dose-dependently decreased, inter-CSD interval was elongated, and propagation velocity was decreased. The total length of time for which CSD continued showed a tendency to decrease, but this was not significant (Figure 4). A small negative deflection of DC potential only at the proximal site but not at the distal site, with a small CBF change or with no CBF change, was detected in seven rats (three rats in the 600 mg/kg group, two rats in the 100 mg/kg group, one rat in the 50 mg/kg group and one rat in the vehicle group) (Figure 5). There was no difference in the CSD-associated elevation of CBF before and after the final administration of TPM (Figure 6).
Average values of CSD occurrence frequency (A), inter-CSD interval (B), total length of time for which CSD continued (C) and CSD propagation velocity (D) in response to KCl application 1 h before the final oral administration of vehicle/TPM in chronically treated rats (t = −1 in Figure 1). The results in TPM-treated groups are not statistically significantly different from the vehicle control. Average values of CSD occurrence frequency (A), inter-CSD interval (B), total length of time for which CSD continued (C) and CSD propagation velocity (D) in response to KCl application 1 h after the final oral administration of vehicle/TPM in chronically treated rats (t = 1 in Figure 1). *p < 0.05, **p < 0.01 significant differences of the TPM-treated groups from the vehicle control. Original recordings of DC potential at the proximal and distal sites and CBF showing an example of non-propagated DC potential deflection. DC potential deflection detected at the proximal site was not propagated to the distal site, as indicated with an arrowhead, and there was no CBF change. Average values of CSD-associated CBF changes in response to KCl application before the final oral administration of drugs (t = −1 in Figure 1) (A) and those in response to KCl application after the final oral administration of drugs (t = 1 in Figure 1) (B). There are no statistically significant differences from the vehicle control.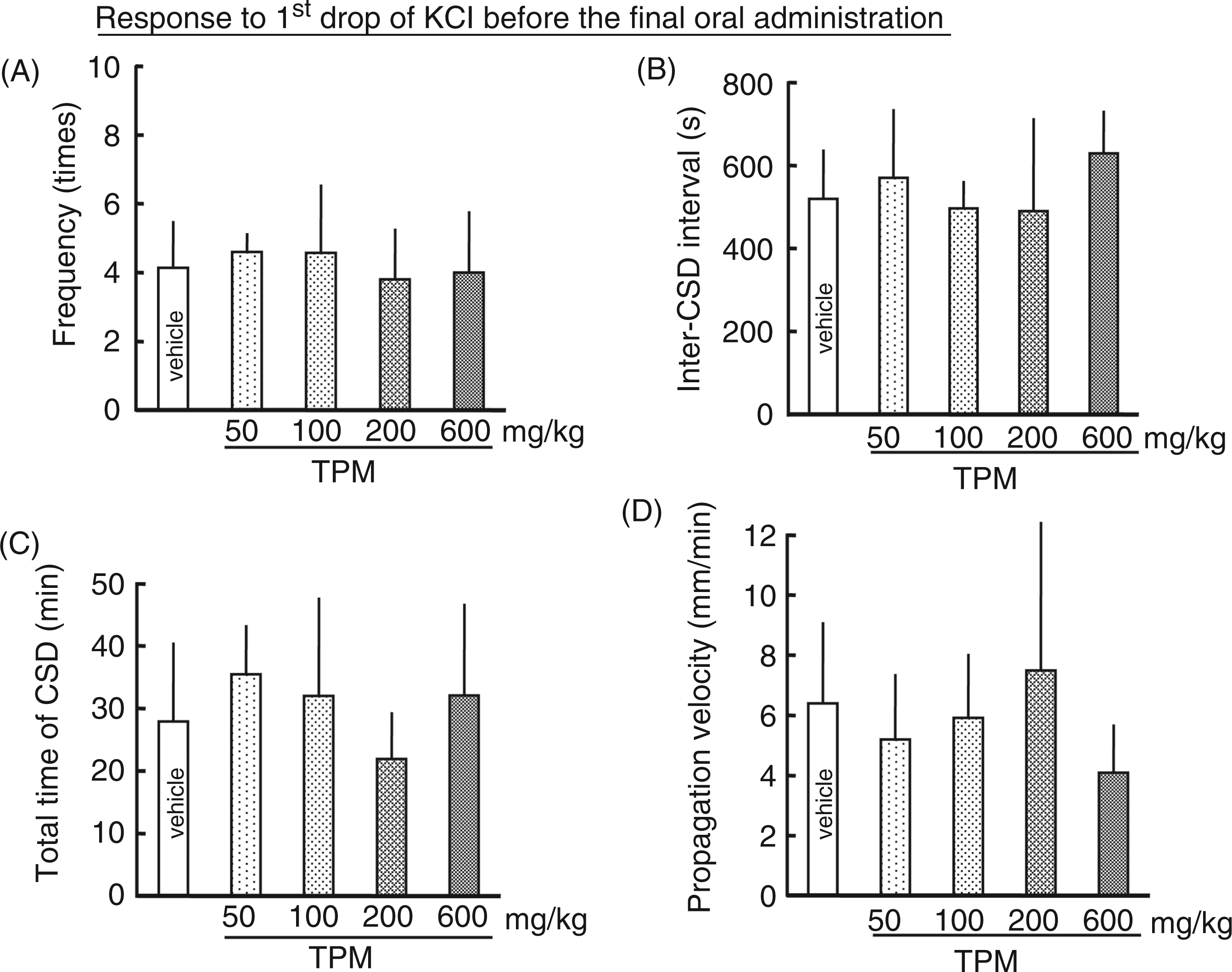


Acute effect of TPM on CSD
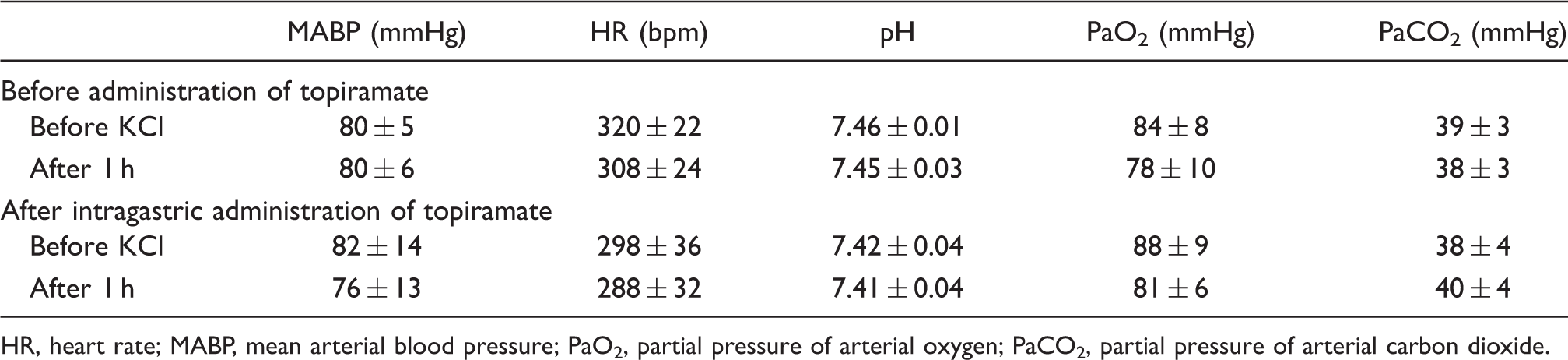
KCl-induced CSD susceptibility (CSD occurrence frequency, inter-CSD interval, propagation velocity and total length of time for which CSD continued) was not significantly influenced, but CSD-associated elevation of CBF was significantly reduced by single administration of TPM (Figure 7). Negative deflection of DC potential only at the proximal site but not at the distal site, with or without a small CBF change, was detected in five rats after TPM administration. Physiological parameters (MABP, HR, arterial pH, partial pressure of arterial oxygen (PaO2) and carbon dioxide (PaCO2)) were within normal ranges and were not significantly changed either by KCl application or by intragastric administration of TPM (Table 2).
Changes of CSD occurrence frequency (A), inter-CSD interval (B), total length of time for which CSD continued (C), CSD propagation velocity (D) and CSD-associated CBF changes (E) from the pre-administration level (before) to the level after single gavage administration of TPM (600 mg/kg). *p < 0.05 significant difference from the pre-administration level. Changes in physiological parameters, arterial pH and arterial blood gas in response to CSD induction with KCl application HR, heart rate; MABP, mean arterial blood pressure; PaO2, partial pressure of arterial oxygen; PaCO2, partial pressure of arterial carbon dioxide.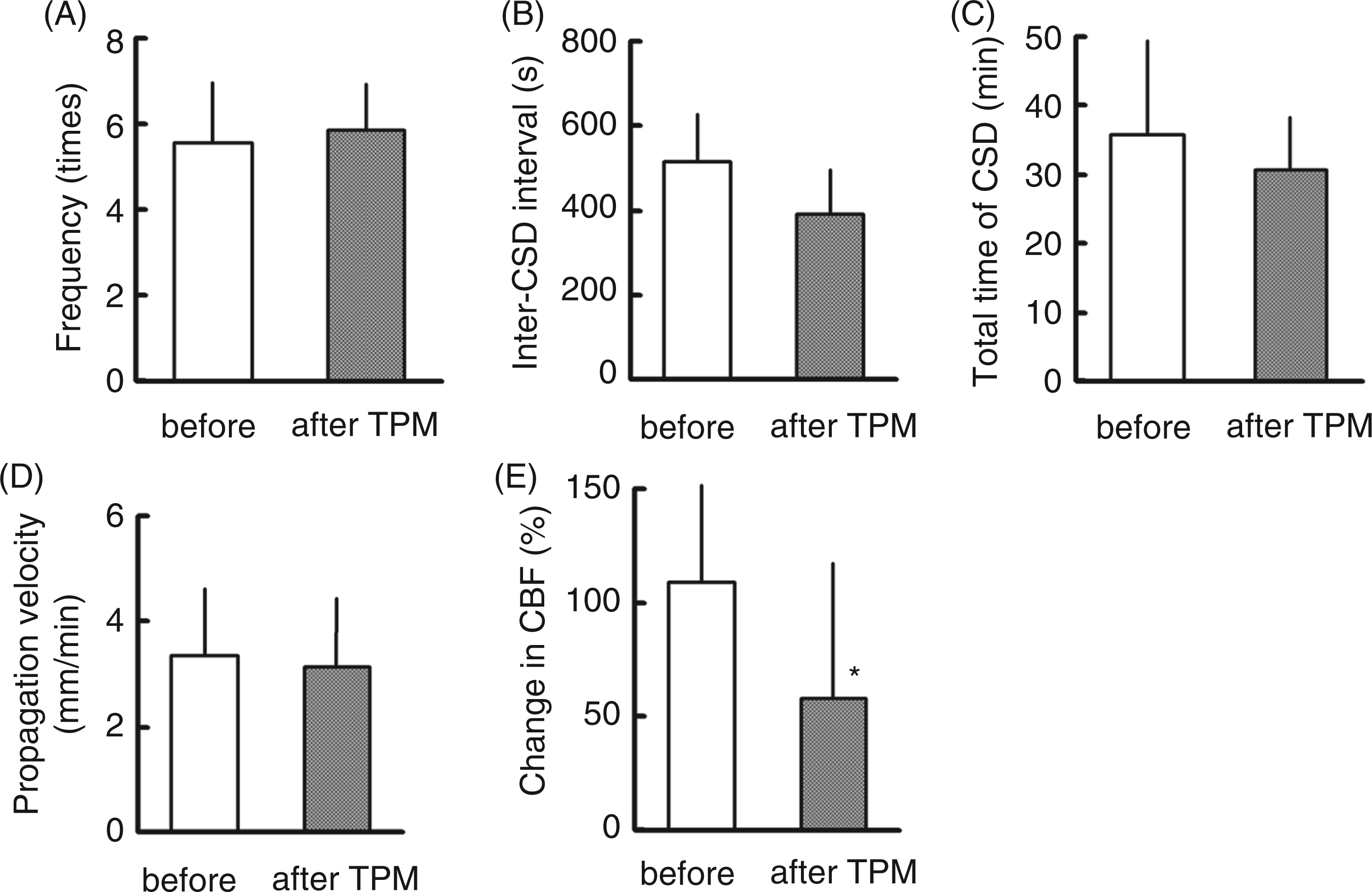
Discussion
In our study, chronic peroral administration of TPM suppressed CSD susceptibility, whereas single administration had little effect on CSD susceptibility. Intraperitoneal administration of TPM for 4 weeks suppressed CSD occurrence frequency induced by KCl application, enhanced the cathodal stimulation threshold and lowered the propagation speed, and longer administration (17 weeks) almost abolished CSD occurrence (23). Thus, oral administration for a longer period would be expected to have a potent effect on CSD susceptibility.
The administered amount and plasma concentration of TPM in the present experiment were much higher than the clinically used levels in human. We demonstrated rapid and stable absorption of orally administered TPM in this study. However, the TPM concentration before the final administration was very low in this experiment, indicating that TPM was largely eliminated within 1 day in rats, and no accumulation effect was observed during daily administration for 6 weeks. The biological half-life (T1/2) in humans is 27–31 h (24). Thus, species difference in TPM turnover rate and/or bioavailability might require a higher dose in rats, and drug accumulation might also be different. We found that CSD occurrence and propagation before the final dose of TPM in the 6-week daily administration protocol were not different from those of the vehicle control. This suggests that there might be a drug threshold of approximately 25 µg/ml in rats. This in turn might be consistent with the observation in the PROMPT trial that cessation of chronic TPM resulted in loss of the prophylactic effect (25), and suggests that monitoring of plasma TPM levels might be important for effective treatment.
Single gavage administration of TPM had no effect on CSD susceptibility in our experiment. It was also reported by others that single intraperitoneal administration did not reduce the number of CSD episodes induced by KCl (23). These facts may suggest that a certain period is required for manifestation of the prophylactic effect of TPM. However, a single dose of intravenous TPM inhibited CSD occurrence induced by needle plunge, but did not influence the CSD propagation speed in anesthetized rats and cats (20). One possible explanation might be a time lag in the transfer of TPM from blood to brain. It is also possible that there is a depot effect due to drug accumulation within a sequestered brain compartment. Although it is possible that a single dose might block propagation in the distal direction as seen in our experiment, a similar phenomenon was also seen in chronically treated rats. CSD seems to be more effectively suppressed by chronic treatment. Although intravenous injection might provide rapid relief of migraine, it may be possible to obtain a long-lasting prophylactic effect in humans by regular intragastric administration, which is a more convenient route.
It was reported that chronic intraperitoneal administration of TPM dose-dependently suppressed CSD susceptibility, though the dosages (40–80 mg/kg/day) were lower than in our study (23). It should be borne in mind that the administration methods were different; intraperitoneal administration in previous studies and oral gavage in our experiment. We found that the maximum plasma level after intraperitoneal administration was approximately three times that after intragastric administration. It seems possible that intragastric administration may result in lower TPM blood levels because of restricted absorption at the gastrointestinal tract, but it may allow the blood level of TPM to be maintained for a longer period, than with intraperitoneal administration. Thus, higher doses may be required in the case of intragastric administration.
CBF elevation related to potassium-evoked CSD was not affected by chronic TPM treatment, whereas it was suppressed by single administration of TPM in our study. This effect of single administration is consistent with that described previously (20). The mechanism of vasodilation associated with CSD is complicated, being influenced by multiple stimuli arising from cerebral arteries and parenchyma, including various neurotransmitters secreted by sensory and parasympathetic nerves (26). TPM diminished neurogenic dural vasodilation by inhibiting the presynaptic release of calcitonin gene-related peptide (CGRP) from trigeminal neurons, but did not act postsynaptically at blood vessels (27). A single dose of TPM might attenuate vasodilation by reducing release of vasodilative substances, such as CGRP, whereas the suppressive response might be attenuated and/or compensated for by other mechanisms in the chronically treated rats.
TPM has been reported to suppress excitatory amino-acid-related synaptic transmission and ion channels (28,29). Conversely, the drug enhances GABAA receptor-mediated inhibitory currents and blocks the GluK1 agonist-mediated suppression of GABA release from interneurons (30). Although it has not been demonstrated that GABA receptors directly modulate CSD susceptibility, it is possible that TPM might suppress CSD occurrence and propagation via interference with excitatory amino-acid-mediated ion channels and/or inhibition of GABA-mediated depolarization.
In our study, we used daily administration of TPM for 6 weeks, so it is possible that the suppressive effect of TPM on CSD involves induction of enzyme/receptor(s) or modulation of gene expression. TPM enhanced GABA release by down-regulation of GABAB autoreceptor expression and up-regulation of astroglial TWIK-related acid-sensitive K+ channel-1 in gerbils (31,32). A neuroprotective effect of TPM was seen as an enhancement of cell survival on exposure of primary neuronal-astroglial cultures to glutamate- and kainate-induced neurotoxicity (33) and blockade of up-regulation of caspase-3 expression in hippocampus of kindled rats (34). These results suggest a possible mechanism of the antiepileptic effects of TPM. A similar mechanism may be involved for migraine. Thus, chronic TPM treatment may induce up- or down-regulation of certain neuronal and/or astroglial proteins, leading to suppression of neuronal activity associated with CSD and resulting in relief of migraine and/or aura.
Another migraine preventive drug, lamotrigine, had a potent suppressive effect on CSD susceptibility (especially CSD occurrence frequency) and c-Fos expression in the cortex, but did not affect propagation velocity (35). Lamotrigine is clinically effective on migraine with aura, but not on migraine without aura. On the other hand, migraine without aura as well as with aura was relieved by TPM in the clinical trial (17). The relieving effect of TPM on migraine might involve not only a decrease in CSD susceptibility, but also other mechanisms.
Conclusion
Chronic treatment with TPM suppressed CSD occurrence and propagation along the cerebral cortex, and is therefore expected to relieve migraine. However, continuous maintenance of a sufficient blood level of TPM appears to be necessary.
Footnotes
Acknowledgements
The authors thank Janssen Pharmaceutical K.K. for measurement of plasma concentration of TPM and generous support.
Funding
This work was supported by Grants-in-Aid for Scientific Research No. 22390182 (to NS) and No. 21591119 (to YT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by Janssen Pharmaceutical K.K.
Conflict of interests
The authors declare that there is no conflict of interest.