
Abstract
Introduction: Calcitonin gene-related peptide (CGRP) is a key molecule in migraine pathogenesis. Intravenous CGRP infusion triggers delayed migraine-like attacks in patients with migraine without aura (MO). In contrast to patients with MO, in prior studies patients with familial hemiplegic migraine (FHM) did not report more migraine-like attacks compared to controls. Whether CGRP triggers migraine in patients with typical (non-hemiplegic) migraine with aura is (MA) unknown. In the present study we examined the migraine inducing effect of CGRP infusion in patients suffering from MA and healthy controls.
Methods: Fourteen patients suffering exclusively from migraine with typical aura (MA) and 11 healthy volunteers received a continuous intravenous infusion of 1.5 µg/min CGRP over 20 minutes. Headache and other migraine symptoms were scored every 10 minutes for one hour and self recorded hourly thereafter and until 13 hours post-infusion.
Results: CGRP infusion induced significantly more delayed headaches in MA patients (12 out of 14) than in controls (2 out of 11) (p = 0.001). Furthermore, significantly more MA patients (57%; 8 out of 14) fulfilled criteria for an experimentally induced migraine attack after CGRP than controls (0%; 0 out of 11) (P = 0.003). Four patients (28%) reported aura symptoms after CGRP infusion.
Conclusion: CGRP triggered migraine-like attacks without aura in patients suffering exclusively from MA. It also triggered a typical aura in 28% of the patients. These data indicate similar neurobiological pathways responsible for triggering migraine headache in MA and MO patients, and suggest differences between MA/MO and FHM.
Keywords
Introduction
Over the last 20 years major advances have occurred in our understanding of the role of calcitonin gene-related peptide (CGRP) in the pathophysiology of migraine (1).
Rosenfeld et al. (2) demonstrated CGRP in the rat trigeminal ganglia and Mason et al. (3) reported in vitro calcium-dependent CGRP release from trigeminal ganglia upon depolarization. A dense supply of CGRP-containing fibers around the cerebral vessels was reported to originate in the trigeminal ganglion (4).
In 1988 Goadsby et al. reported that thermocoagulation of the trigeminal ganglion leads to CGRP release into the extracerebral circulation of humans (5). This was the first report of CGRP involvement in the trigeminovascular reflex. The importance of CGRP in migraine became firmly established when infusion of CGRP caused migraine-like attacks in patients with migraine without aura (MO) (6) and CGRP-receptor antagonism was established as an effective treatment of migraine attacks (7–9).
Despite new insights (10), the exact pathways involved in CGRP-induced migraine attacks and mechanisms of action of CGRP antagonists are largely unknown. Furthermore, the importance of CGRP across the migraine spectrum—ranging from MO, migraine with aura (MA) to familial hemiplegic migraine (FHM)—is only now being clarified. Recently, we examined the effect of CGRP provocation in the rare subtype of MA called familial hemiplegic migraine types 1 and 2 (11). In contrast to patients with MO, FHM patients did not report more migraine-like attacks compared to controls. These data suggest distinct molecular mechanisms underlying FHM and MO and raised the question whether FHM should be considered as a part of the migraine spectrum. Currently, FHM and MA are both classified under the heading “migraine with aura” (12) and the underlying mechanism of the aura both in FHM and in MA is thought to be a cortical spreading depression (CSD) (13–16). The present study addresses whether these disorders share migraine pain mechanisms and whether systemic administration of CGRP in MA patients would fail to induce migraine-like attacks similar to those experienced by FHM patients. To test this hypothesis we examined the migraine-inducing effect of CGRP infusion in patients suffering from migraine with typical aura and healthy controls.
Design and methods
Clinical characteristics of spontaneous migraine with aura attacks

V = visual; A = aphasic; S = sensory; Occ = occasionally; VAS = visual analogue scale.
We included 11 healthy volunteers for the control group (4 women, 7 men, mean age 36 years [range 22–65]). The controls had no personal or family history of migraine. Exclusion criteria were: any other headache disorder, except episodic tension-type headache fewer than five days per month; daily medication apart from oral contraceptives; serious somatic or psychiatric disease. The Ethics Committee of the County of Copenhagen (HA2007-0050) approved the study, which was undertaken in accordance with the Helsinki Declaration of 1964, as revised in Edinburgh in 2000. All subjects gave informed consent to participate in the study. The study was registered at ClinicalTrials.gov (NCT00687947).
Experimental design
The study design was non-randomized, balanced, controlled and single-blinded. All subjects received a continuous intravenous infusion of 1.5 µg/min CGRP over 20 minutes. Human αCGRP was purchased from Clinalfa AG, Läufelfingen, Switzerland. The subjects were informed that CGRP might induce headache in some individuals, but the timing or the type of headache was not discussed. None of the subjects had previously participated in provocation studies.
All subjects reported headache-free to the laboratory. All procedures were performed in a quiet room with the subjects placed in the supine position. A venous catheter (Venflon®) was inserted into an antecubital vein and after 30 minutes of rest, the infusion was started, using an infusion pump (Braun Perfusor, Melsungen, Germany). Headache intensity, adverse events and vital signs were recorded at T−10, and then every 10 minutes until 60 minutes after start of infusion. The subjects were discharged from the hospital after finishing the measurements and were asked to complete a headache diary every hour until 13 hours after start of infusion and a questionnaire to report headache and migraine in the 24-hour period after infusion. The diary and questionnaire included headache characteristics and accompanying symptoms according to the IHS (12), any rescue medication taken and adverse events. In addition, all subjects were asked to report whether the delayed headache mimicked their usual headache and migraine patients also reported if headache mimicked their usual migraine attack. Subjects were allowed to treat headache with over-the-counter rescue medication or their usual migraine treatment.
Headache intensity and migraine definition
Headache intensity was recorded on a verbal rating scale (VRS) from 0 to 10 (0, no headache; 1, a very mild headache, including a feeling of pressing or throbbing; 5, moderate headache; 10, worst imaginable headache) (17). We also recorded headache characteristics and associated symptoms to determine if the headache fulfilled international diagnostic criteria for any type of migraine (12). Most attacks of MO develop in a matter of hours and often go through a phase where they phenomenologically fulfill the criteria for tension-type headache before the headache gets worse, becomes unilateral and has the associated symptoms required for migraine. For this reason attacks aborted by migraine-specific treatment were accepted in the new criteria for chronic migraine (18). Patients in experimental provocation studies cannot be denied treatment of the induced attacks and often treat before all migraine criteria are fulfilled. We have therefore used the following criteria for an experimentally induced migraine attack: Migraine attacks after infusion of an experimental drug fulfilling criteria C and D for migraine without aura or headache described as mimicking usual migraine attack and treated with a triptan (Criteria C: moderate to severe pain intensity is considered 4 ≤ on VRS scale, criteria D: during headache at least one of the following: either nausea and/or vomiting or photophobia and/or phonophobia).
Data analysis and statistical methods
All values are presented as mean ± standard deviation (SD), unless otherwise stated. We defined the period from 0 to 60 minutes after the start of infusion (when the subjects were still in the clinic) as the infusion phase and the period from 1 hour to 13 hours after the start of infusion as the post-infusion phase. Baseline was defined as −10 minutes before the start of infusion.
Calculation of sample size was based on the detection of a difference between the MA group and controls reporting CGRP-induced migraine attack within the 13 hours following infusion (1 hour–13 hours), at 5% significance with 80% power. We assumed that CGRP would induce a delayed migraine-like attack in 50% of MA patients, as reported previously in MO patients (6), and migraine-like headache in less than 10% of healthy controls (19). We estimated that 11 subjects should be included in each group (20).
The area under the curve (AUC) was used as summary measure for analyzing differences between the groups and was calculated according to the trapezium rule (21).
The primary endpoints were differences in incidence of migraine-like attacks (1–13 hours) between MA patients and controls. Additional endpoints were differences in incidence for headache (0–1 hour and 1–13 hours) and AUC for headache score (AUCheadache 0–60 min and AUCheadache 1–13 h) between MA and controls.
The incidence of migraine-like attacks and adverse events between the groups was compared with Fisher's exact test. All other statistical analysis was performed using an unpaired, two-way t-test, except headache scores, where data are presented as medians and quartiles and tested with the Mann-Whitney test. All analyses were performed with SPSS for Windows 17.0 (Chicago, IL, USA). Five percent (p < .05) was chosen as the level of significance.
Vital signs
Heart rate and blood pressure were measured every 10 minutes using an auto-inflatable cuff (ProPac Encore®; Welch Allyn Protocol). ECG (Cardiofax V; Nihon-Koden, Shinju-ku, Tokyo, Japan) was monitored on a LCD screen and recorded on paper every 10 minutes.
Results
All 25 participants completed the study. They were headache-free at baseline. We found no baseline difference in heart rate and blood pressure between the groups (p > .05).
Migraine-like attacks after CGRP
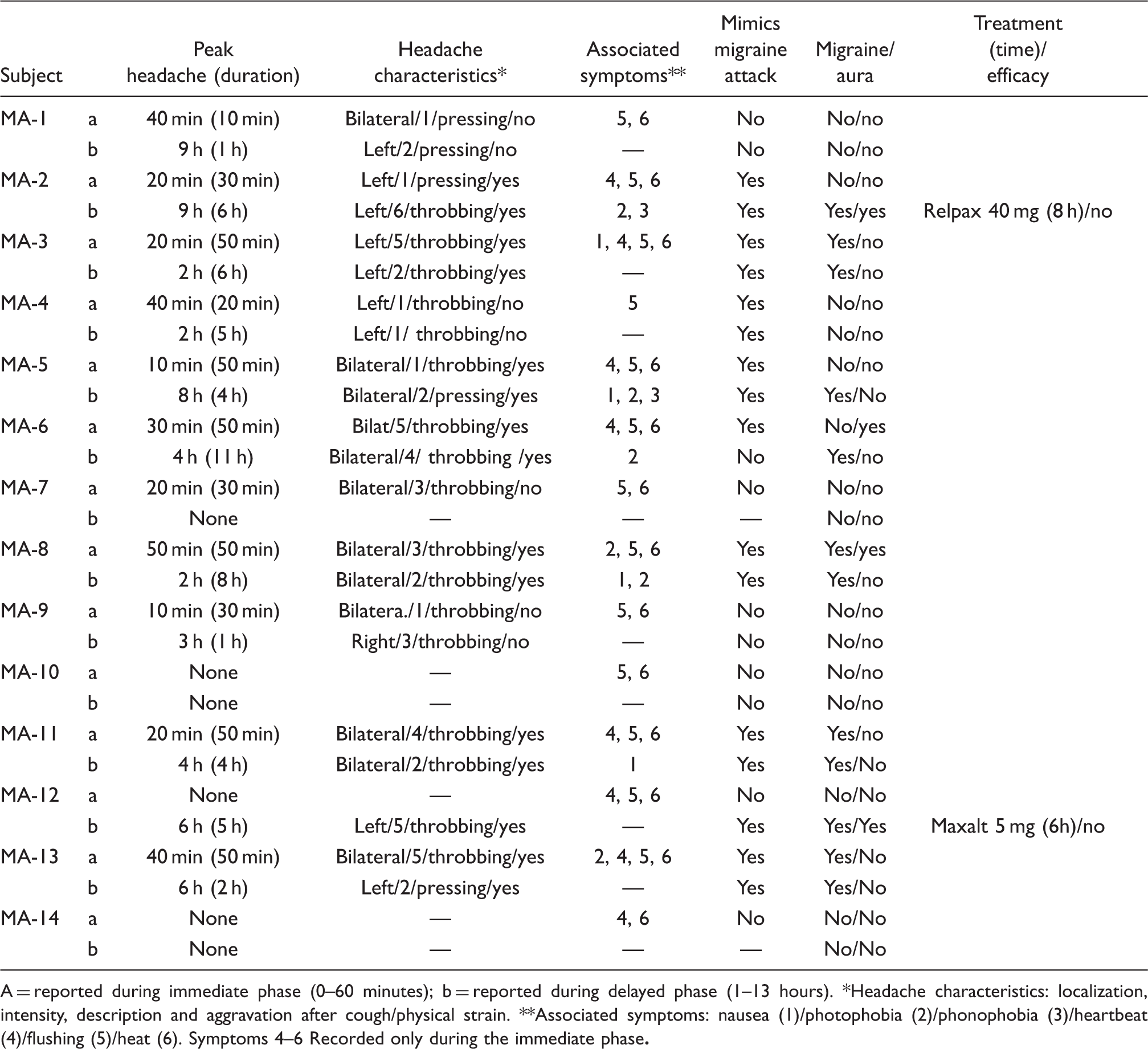
In total, 12 migraine patients (86%) experienced headache after CGRP infusion (0–13 hours), and 8 (57%) of these also reported migraine-like attacks (Figure 1; Table 2). In the post-infusion phase (1–13 hours), significantly more MA patients (57%; 8 out of 14) reported migraine-like attacks than controls (0%; 0 out of 11) (p = .003) (Table 2). Two patients took rescue medication (Table 3).
Median (thick lines) and individual (thin lines) headache scores on a verbal rating scale (VRS) during immediate (0–60 min) and delayed phases (1–13 h) after start of the CGRP infusion in 14 patients with MA (black) compared to 11 controls (white). MA patients and controls reporting headache and migraine-like attacks after CGRP*
MA = migraine with aura; CGRP = calcitonin gene-related peptide. *Groups compared with Fisher's exact test. Headache characteristics after CGRP infusion in MA patients A = reported during immediate phase (0–60 minutes); b = reported during delayed phase (1–13 hours). *Headache characteristics: localization, intensity, description and aggravation after cough/physical strain. **Associated symptoms: nausea (1)/photophobia (2)/phonophobia (3)/heartbeat (4)/flushing (5)/heat (6). Symptoms 4–6 Recorded only during the immediate phase

Headache intensity
The area under the headache curve in the infusion phase (AUCheadache 0–60 min) tended to be larger in MA patients than in controls (p = .066). Peak median headache (1.0) in the MA group occurred 20 minutes after start of the CGRP infusion, while the median headache was 0 at all time points in the control group (Figure 1). The area under the headache curve in the post-infusion phase (AUCheadache 1 h–13 h) was larger in the MA group than in controls (p = .003). Peak median headache (0.5) occurred 9 hours after start of the CGRP infusion, while the median headache was 0 at all time points in the control group (Figure 1).
Overall headache induction
The incidence of reported headache in the infusion phase was not different between the two groups (11/14 MA patients vs. 7/11 controls) (p = .65), but more MA patients than controls reported delayed headache in the post-infusion phase (12/14 MA patients vs. 2/11 controls) (p = .001) (Table 2).
Aura
Four patients (28%) reported aura symptoms and migraine-like headaches (Table 3). Apart from the duration of aura attack in patient 2, all patients described the symptoms as being similar to those of a spontaneous MA attack.
Patient 2 reported left-sided visual aura with fortification spectra two hours after start of the infusion. The aura lasted for 10 hours and continued throughout the ensuing migraine-like headache.
Patient 6 reported bilateral flickering light spots starting 20 minutes after start of infusion, and lasting for approximately 10 minutes.
Patient 8 reported left-sided homonymous scotoma with the onset of headache, 10 minutes after start of the infusion. The symptoms lasted approximately 10 minutes.
Patient 12 reported visual and sensory aura starting concomitantly with the migraine-like headache six hours after start of infusion. The visual symptoms were right-sided homonymous scotoma, and the sensory symptoms were tingling paresthesia in the fingers of the right arm and right side of the face. The symptoms lasted approximately 15 minutes.
Mean arterial blood pressure and heart rate
There were no differences in mean arterial blood pressure (MAP) (AUCMAP, p = .95) or heart rate (HR) (AUCHR, p = .20) between patients and controls. Mean percentage changes from baseline are shown in Figure 2.
Changes in mean arterial blood pressure (MAP) (squares) and heart rate (HR) (triangles) during and after infusion of CGRP (MA: black, Controls: white). The peak decrease in MAP was 6.8% ± 1.7 (mean ± SEM) in patients and 10.0% ± 2.4 in controls and occurred 20 min after start of the infusion. The maximum increase in heart rate was 25.7% ± 2.9 in patients and 13.8% ± 2.0 in controls and occurred 20 min after start of the infusion. We found no statistical difference in the area under the curve for MAP or heart rate between patients and controls.
Adverse events
All reported adverse events are well known after CGRP infusion: flushing, palpitation and heat sensation (Table 3). None of participants reported severe hypotension, as previously reported by Lassen et al. (6).
Discussion
The major outcome of the present study is that systemic administration of CGRP induces more migraine-like attacks in MA patients than in controls.
Migraine-like attacks after CGRP
Whether MA and MO represent two points on a continuum (22–25) and whether silent CSD occurs in MO has been debated for decades (16,26,27). From the nosographic viewpoint, MO and MA display a similar headache phase (12) and treatment response to specific anti-migraine drugs (28,29). This suggests that MA and MO share some basic pain mechanisms. Using the CGRP provocation model of migraine, it is possible to examine pathophysiological pathways involved in migraine induction and to define whether different migraine subtypes share these pathways.
Lassen et al. (6) examined the effect of CGRP infusion in 12 patients with MO in a double-blind, placebo-controlled, cross-over study. The authors reported that three out of nine patients experienced delayed migraine according to the stringent IHS criteria, versus one patient after placebo. But if we carefully examine the clinical characteristics presented in the paper by Lassen et al. (6), at least three other patients reported migrainous features such as nausea, aggravation by physical activity, photophobia and unilateral (similar to spontaneous attacks) headache treated with sumatriptan. By adding these patients to responders, the percentage of reported migraine-like attacks would increase to 50%. Therefore, we powered our study based on a 50% responder rate in MO and we expected a similar responders rate in MA patients. In the present study 57% of MA patients reported delayed migraine-like attacks compared to none in the control group. These data indicate similar CGRP migraine-triggering mechanisms in MO and MA. Interestingly, similar results were previously reported using glyceryl trinitrate (GTN) model of migraine. Eighty per cent of MO patients (30,31) and 50% of MA patients (32) reported migraine-like attacks after GTN. However, CGRP (11) and GTN (33,34) failed to induce more migraine-like attacks in patients with FHM types 1 and 2 compared to controls. Based on these data, we suggest that neurobiological pathways responsible for triggering migraine headache in MO and MA patients are probably similar, but might be distinct from FHM.
Migraine aura after CGRP
MA is likely to be caused by CSD (14,15,35). The role of CGRP in CSD animal models has not yet been determined. At the molecular level it has been shown that infusion of the neuropeptide endothelin-1 is able to cause CSD (36), probably via stimulation of phospholipase C (37). Interestingly, CGRP acts in part via the same mechanism (38). It would be interesting to examine the possibly increased susceptibility to CSD in CGRP-sensitized transgenic mice (30).
Attempts have been made to trigger aura in migraineurs using human models of migraine. Using GTN, Christiansen et al. (32) demonstrated that 50% of the patients suffering exclusively from MA developed migraine headache with associated symptoms but none of them developed migraine aura. Afridi et al. (30) reported that one out of 21 patients with MA had an aura triggered on two separate occasions by GTN. To our knowledge the present study is the first to report aura symptoms after CGRP infusion. We found that 28% (4 out of 14) of patients developed aura symptoms associated with migraine headache. All patients described the symptoms as being similar to their spontaneous MA attacks. Interestingly, CGRP infusion did not induce an aura in any FHM type 1 or 2 patients (11). This might be due to a difference in attack frequency: MA patients in our study had more frequent aura attacks (range 2–24 per year, Table 1) than FHM patients (range 1–10 per year), as reported in our previous studies (11). In MO, GTN triggering of attacks was, however, independent of usual attack frequency (40). At present we cannot exclude that aura attacks were due to experimental stress, and it would be interesting to re-challenge these patients with CGRP infusion.
How might CGRP induce migraine?
Given that CGRP is a strong dilator of cephalic arteries (41) involved in the trigeminovascular reflex (5) and that systemic CGRP does not cross the blood brain barrier in humans (19), the most obvious site of CGRP-induced nociception or migraine pain would be the extracerebral perivascular space. However, animal studies have provided conflicting data. In one study, CGRP-induced dilatation of meningeal blood vessels caused a sensitization of central trigeminal neurons and a facilitation of facial sensory processing, which was blocked by activation of pre-synaptic 5-HT1B/1D receptors (42). In contrast, topical administration and intravenous infusion of rat α-CGRP in anesthetized rats caused a significant increase in dural blood flow but no activation or sensitization of meningeal nociceptors (43).
In MO patients CGRP infusion caused a modest, if not questionable, dilatation of the middle cerebral artery (7.5%) (44) and marked dilatation of superficial temporal and radial arteries in healthy subjects during the immediate phase (19). At the intracellular level it is known that CGRP activates adenyl cyclase, increases cyclic adenosine monophosphate (cAMP) concentration and thus dilates blood vessels (45). This pathway may play an important role in CGRP-induced migraine attacks. In support, cilostazol, a PDE 3 inhibitor known to upregulate cAMP levels, was found to cause severe migraine-like headaches in healthy subjects (46).
Concluding remarks
The present study demonstrates that CGRP triggers migraine-like attacks without aura in 57% of patients suffering exclusively from MA and typical aura attacks in 28% of these patients. Migraine pain–inducing data are similar to those reported in MO patients and contrast with findings in FHM patients.
Footnotes
Acknowledgements
The authors thank all participating patients and healthy volunteers. This work was supported by grants from the University of Copenhagen, the Danish Headache Society and through the Lundbeck Foundation Center for Neurovascular Signalling (LUCENS).