
Abstract
Objective
Probable migraine is a common, disabling migraine subtype fulfilling all but one of the diagnostic criteria for migraine. This study was conducted to evaluate the efficacy and tolerability of sumatriptan/naproxen sodium for the acute treatment of probable migraine without aura.
Methods
Patients treated a headache of probable migraine without aura when pain was moderate or severe with sumatriptan/naproxen sodium (n = 222 intent-to-treat (ITT)) or placebo (n = 221 ITT/complete case analysis a ) in this randomized, double-blind, parallel-group study.
Results
Sumatriptan/naproxen sodium was more effective than placebo with respect to the co-primary efficacy endpoints two-hour pain-free response (29% sumatriptan/naproxen sodium vs 11% placebo, p < 0.001) and two- to 24-hour sustained pain-free response (24% sumatriptan/naproxen sodium vs 9% placebo, p < 0.001). Sumatriptan/naproxen sodium was significantly more effective than placebo with respect to the secondary efficacy endpoints of pain-free response four hours postdose (p < 0.001), pain-free response maintained one to two hours postdose (p = 0.034) and two to four hours postdose (p < 0.001), headache relief four hours postdose (p < 0.001), headache relief maintained two to four hours postdose (p = 0.015), sustained headache relief two through 24 hours postdose (p = 0.002), and rescue medication use (p < 0.001); but not productivity scores. The most common adverse events were dizziness (4% sumatriptan/naproxen sodium,<1% placebo), dry mouth (2% sumatriptan/naproxen sodium, <1% placebo), and nausea (2% sumatriptan/naproxen sodium, <1% placebo).
Conclusion
Sumatriptan/naproxen sodium is effective in the acute treatment of probable migraine as demonstrated by higher rates of freedom from pain and restoration of function.
Introduction
In the International Classification of Headache Disorders, second edition (ICHD-II), the term probable migraine is applied to headache syndromes meeting all except one of the diagnostic criteria for migraine with/without aura (1). This category is intended for patients who do not fully meet diagnostic criteria for migraine but are judged to have it. This group is important in clinical practice because probable migraine is common, with a prevalence of 3%–10%; and it disrupts quality of life and impairs function (2–7). Like migraine, probable migraine is under-recognized—often misdiagnosed as another headache type such as sinus or tension type—and undertreated (8–10). Treatment and treatment guidelines are limited by the lack of Class I evidence for effective treatment. Therefore, it is important to design clinical trials to provide Class I evidence to inform physicians who are most likely to treat patients with probable migraine, i.e. primary care/family practice.
Current treatment is based on the assumption that the pathophysiology and treatment response profile of probable migraine are similar to those of migraine (1,11–13). In this context, sumatriptan/naproxen sodium, a combination tablet containing sumatriptan 85 mg formulated with a rapid-release formulation (RT Technology) and naproxen sodium 500 mg, is a promising treatment for probable migraine. Sumatriptan/naproxen sodium targets vascular, neuronal, and inflammatory processes and is more effective than its individual components with proven efficacy as acute treatment for moderate/severe migraine, as early intervention for migraine, and in migraine subpopulations (14–19). The study reported herein was conducted to evaluate the efficacy and tolerability of sumatriptan/naproxen sodium in the acute treatment of a moderate-to-severe probable migraine without aura.
Methods
Patients
Males and nonpregnant, nonlactating females 18 to 65 years old were eligible for the study if they had a ≥6-month history of probable migraine without aura defined by ICHD-II criteria (1); had, during the six months preceding the screening visit, an average of one to six episodes/month of probable migraine, typically with moderate or severe headache that lasted four to 72 hours if untreated or unsuccessfully treated; had ≥20 headache-free days/month in each of the three months before the screening visit; and could distinguish probable migraine episodes from other types of headache. Females had to be physiologically incapable of becoming pregnant or, if they could become pregnant, to agree to practice adequate contraception during the study.
Patients were excluded if they had headaches (any type) for >10 days/month in any of the three months before the screening visit; had used any 5HT1 receptor agonist, an ergotamine-containing migraine medication, an ergot derivative, or methysergide; were taking a medication prescribed for headache prophylaxis; had migraine (including basilar, hemiplegic, or ophthalmoplegic migraine) or met ICHD-II criteria (1) for migraine with or without aura at the screening visit; had used opioids or barbiturates for headache or pain on an average of ≥4 days/month over the six months before the screening visit; had uncontrolled hypertension at the screening visit (sitting diastolic ≥90 mmHg, systolic ≥140 mmHg) or were taking an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker; had confirmed or suspected cardiovascular or cerebrovascular disease; had a history of cardiac arrhythmias requiring medication or of clinically significant electrocardiogram (ECG) abnormalities that, in the investigator’s opinion, contraindicated study participation; or were taking a monoamine oxidase inhibitor or had taken any monoamine oxidase inhibitor or St. John’s Wort within two weeks before the screening visit. All patients provided written informed consent prior to study participation. Patients were thus prophylaxis free and triptan naive and did not have complicating migraine characteristics.
Patients could be recruited into the study by using chart review, advertising, or normal patient flow into the clinical practice.
Procedures
The protocol for this randomized, double-blind, placebo-controlled, parallel-group study (GlaxoSmithKline protocol TXA107563; clinicaltrials.gov number NCT00387881) was approved by local or regional institutional review boards for each of the 53 United States (US) study sites. The study was conducted from September 14, 2006 to February 27, 2008. The study consisted of a screening visit followed by outpatient treatment of a single probable migraine episode and a follow-up visit occurring three to seven days after treatment. At screening, patients meeting eligibility criteria were randomized 1:1 to take a single tablet of sumatriptan/naproxen sodium (sumatriptan succinate equivalent to 85 mg sumatriptan and naproxen sodium 500 mg) or placebo and were instructed to treat a probable migraine attack with study medication when pain intensity was moderate or severe based on a four-point pain scale (0 = no pain; 1 = mild pain; 2 = moderate pain; 3 = severe pain). Patients were aided by an electronic diary (e-diary) in determining whether a headache qualified as a probable migraine.
Beginning two hours postdose, patients were permitted to take one additional sumatriptan/naproxen sodium tablet or other permitted therapy as open-label rescue medication. Examples of other permitted medications included: naproxen (if used, the recommended dose was 220 mg × two tablets, then one additional tablet (220 mg) six hours later if needed, not to exceed three tablets in a 24-hour period); sumatriptan succinate tablets (if used, the recommended dose was 100 mg maximum, in a 24-hour period); and any other medications commonly used by the subject to treat headache, as long as it was not on the nonpermitted list (e.g. acetaminophen or ibuprofen). Prohibited medications included monoamine oxidase inhibitors at any time during the study; 5HT1B/D agonists or long-acting nonsteroidal anti-inflammatory drug-containing (NSAID-containing) compounds within 24 hours before taking study medication; 5HT1B/D agonists or long-acting NSAID-containing compounds other than sumatriptan/naproxen sodium, sumatriptan, or naproxen sodium within 24 hours after taking study medication (except aspirin ≤325 mg/day for cardiovascular prophylaxis); analgesics containing morphine, codeine, a barbiturate, or an opioid derivative from 24 hours before through 24 hours after taking study medication; or short-acting NSAID-containing compounds or other analgesics or antiemetics from six hours before through two hours after taking study medication. Adverse events were captured at the follow-up visit based on patient recall, using an open-ended verbal question to avoid introduction of bias.
Sample size and data analysis
The study was initially planned to randomize 620 patients to obtain 550 (assumed 88% of randomized patients will treat a probable migraine attack within the allowed three months) treated patients, of whom approximately 390 patients (195 patients/treatment arm) would treat a probable migraine (assumed that 70% of patients who treat a headache will treat a headache classified as probable migraine).
Based on a blinded review of the e-diary data, the proportion of patients treating a probable migraine headache in three months was smaller than expected; therefore the number of patients to be randomized was increased to 680 patients. Sample size calculations were based on an assumed comparison of 30% vs 15% for two-hour pain-free rates, and 25% vs 13% for two- to 24-hour sustained pain-free rates. The estimated overall combined power was 92%, with an alpha of 5%. An in-house computer-generated system was used to randomize the subjects into the two treatment groups, using a block size of four. This system keeps all individuals blinded to study treatment until the database is locked. A separate, yet connected, in-house interactive-voice-response-system was used to dispense blinded study drug to the individual sites.
Baseline demographics and clinical characteristics (including Headache Impact Test (HIT)-6 scores (20)), efficacy data, and health outcomes data (i.e. functional ability, productivity, satisfaction) were summarized for the intent-to-treat (ITT) population, defined as randomized patients who treated a headache with study medication and completed ≥1 post-baseline efficacy assessment. Safety and tolerability data were summarized for the safety population, defined as randomized patients who took randomized study medication or open-label rescue medication with sumatriptan/naproxen sodium.
Efficacy
The primary efficacy analysis was to be conducted in patients who eligibly treated a headache with study medication and provided post-treatment efficacy data at least one point in time. The protocol referred to this as an ITT analysis as was cutomary at the time the study was planned. Current Consolidated Standards of Reporting Trials (CONSORT) guidelines suggest the term “complete case analysis” for this group. Efficacy data were summarized from patients’ e-diary records of headache pain (none, mild, moderate, severe) at designated time points through 24 hours following dosing with study medication. The co-primary efficacy endpoints were the percentage of patients with pain-free response (i.e. post-treatment pain severity of none with no use of rescue medication) two hours after treatment and the percentage of patients with two- to 24-hour sustained pain-free response (i.e. no pain from two to 24 hours postdose with no use of rescue medication). Secondary efficacy endpoints included the percentages of patients with 1) pain-free response four, one, and 0.5 hours postdose; 2) sustained pain relief two to 24 hours postdose; 3) pain relief at four, two, one, and 0.5 hours; 4) incidence of headache pain rescue medication use up to 24 hours; 5) intermediate sustained pain relief defined as headache relief maintained from two to four hours and one to two hours postdose; 6) intermediate sustained pain free defined as pain-free response maintained from two to four hours and one to two hours postdose; 7) incidence of headache-associated neck pain at four and two hours; 8) incidence of headache-associated sinus pain at four and two hours; 9) incidence of headache-associated photophobia at four and two hours; 10) incidence of headache-associated phonophobia at four and two hours; and 11) incidence of headache-associated nausea at four and two hours.
Hierarchical (sequential) testing was used for the efficacy endpoints to control the Type I error rate for multiple comparisons. When findings for an endpoint were statistically significant, the next listed endpoint was tested along with any other timepoints (sequentially) for the significant endpoint. Once nonsignificance was observed, any further p value comparisons in the testing sequence, even if less than 0.05, were not considered statistically significant. For endpoints that were measured at multiple timepoints, only significance of the initial timepoint was required to test the next endpoint. For all secondary endpoints, if rescue medication was used prior to the time point, the outcome was not achieved.
Health outcomes
Functional ability was assessed through each patient’s e-diary records using the Clinical Disability Questionnaire (CDQ). The CDQ is a single-item questionnaire that uses a five-point impairment response scale (normal, mildly impaired, moderately impaired, severely impaired, required bed rest) to assess ability to function at a certain timepoint. The CDQ was administered at initial dosing (baseline) and at two and four hours postdose. Treatments were compared using the Cochran-Mantel-Haenszel (CMH) test.
Productivity was assessed based on a short questionnaire derived from the Work Productivity Assessment Inventory (WPAI). This six-item assessment was completed 24 hours after dosing with study medication. The productivity items were designed to measure the impact of a migraine on paid work as well as activities other than paid work. This assessment includes the number of hours missed (absenteeism), the number of hours worked with symptoms (presenteeism), and the effectiveness while completing an activity with symptoms during the 24 hours after taking study medication (21–23). Responses were summarized to yield a measure of total disability time:


Total disability time, lost workplace productivity, and lost activity time were compared between treatment groups using Wilcoxon Rank sum tests.
Satisfaction with medication was assessed at the screening visit (prior medication) and at 24 hours after taking study medication using the Revised Patient Perception of Migraine Questionnaire (PPMQ-R) (24,25). The PPMQ-R used in this study contains 30 items comprising four subscales: efficacy (11 items), functionality (4 items), ease of use (2 items) (each scored from 1 [very dissatisfied] to 7 [very satisfied]), and tolerability (10 items) (scored from 1 [extremely bothersome] to 5 [not at all bothersome]). In addition, the PPMQ-R contained three global satisfaction items—one each assessing overall satisfaction, medication effectiveness satisfaction, and side effect satisfaction—scored on a scale ranging from 1 (very dissatisfied) to 7 (very satisfied). Two PPMQ-R items on cost were not included in the study questionnaire because patients did not pay for study medication.
Scores for each of the PPMQ-R subscales were calculated from item scores while the global scores were reported separately. Each subscale score ranges from 0 to 100, with higher scores indicating greater satisfaction or tolerability. A total score, a composite of the subscale scores for efficacy, functionality, and ease of use, were also computed. Treatment groups were compared using analysis of variance (ANOVA). Previous research suggests that a five-point difference in the efficacy, functionality, and ease of use subscale scores and the total score is clinically meaningful (24,25). Global satisfaction items were summarized as the percentage of patients satisfied/very satisfied, and these treatment groups were compared with the CMH test.
Safety and tolerability
The main safety and tolerability measures were the incidences of adverse events, drug-related adverse events, and premature withdrawal from the study because of adverse events. Adverse events were captured by the investigator or designee at the final visit based on the patient’s response to an open-ended question. An adverse event was defined as any untoward medical occurrence temporally associated with the use of study medication regardless of its suspected cause. A drug-related adverse event was an adverse event that the investigator considered to be at least possibly caused by study medication. Safety and tolerability data were summarized with descriptive statistics.
Results
Patients
The number of patients randomized to treatment was 679 (n = 345 sumatriptan/naproxen sodium, n = 334 placebo) (Figure 1). No patient prematurely withdrew because of an adverse event. The safety population comprised the 500 patients (n = 254 sumatriptan/naproxen sodium, n = 246 placebo) who took study medication or open-label sumatriptan/naproxen sodium as rescue medication. The ITT population comprised the 443 randomized patients (n = 222 sumatriptan/naproxen sodium, n = 221 placebo) who treated at least one headache with study medication and had at least one post-baseline efficacy assessment.
Patient disposition and analysis population.
Demographics and baseline clinical characteristics (intent-to-treat population).

NSAIDs: nonsteroidal anti-inflammatory drugs; HIT-6: Headache Impact Test-6.
Efficacy
Sumatriptan/naproxen sodium was more effective than placebo with respect to both of the co-primary efficacy endpoints: two-hour pain-free response (29% sumatriptan/naproxen sodium vs 11% placebo, p < 0.001) and two- to 24-hour sustained pain-free response (24% sumatriptan/naproxen sodium vs 9% placebo, p < 0.001) (Figure 2). Sumatriptan/naproxen sodium was also more effective than placebo with respect to the secondary efficacy endpoints of pain-free response four hours postdose (p < 0.001), pain-free response maintained one to two hours postdose (p = 0.034) and two to four hours postdose (p < 0.001), headache relief four hours postdose (p < 0.001), headache relief maintained two to four hours postdose (p = 0.015), sustained headache relief two through 24 hours postdose (p = 0.002), and rescue medication use (p < 0.001). Sumatriptan/naproxen sodium did not significantly differ from placebo with respect to pain-free response 0.5 and one hour postdose; headache relief 0.5, one, or two hours postdose; or headache relief maintained one to two hours postdose (Table 2). Sumatriptan/naproxen sodium was also significantly more effective than placebo in traditional (photophobia, phonophobia, nausea) and nontraditional associated symptoms (neck pain and sinus pain) at four hours (p < 0.05) but not at two hours (Table 3).
Results for the co-primary endpoints: Percentages of patients with pain-free response two hours postdose and with sustained pain-free response two to 24 hours postdose. Results for secondary efficacy endpoints
a
and health outcomes endpoints. See Figure 2 for results for primary endpoints. bp < 0.001 vs placebo. cp < 0.05 vs placebo. Results for traditional and nontraditional associated symptoms. p < 0.05 vs placebo. bp ≤ 0.001 vs placebo.


Health outcomes
Functional ability and productivity
At pre-dose baseline, the percentage of patients reporting “normal” functioning did not significantly differ between the sumatriptan/naproxen sodium group (6%) and the placebo group (11%). However, the percentage of patients reporting “normal” functioning at both two hours postdose (p = 0.036) and four hours postdose (p < 0.001) was greater with sumatriptan/naproxen sodium than placebo (Table 2). Productivity ratings in this cohort did not differ between the sumatriptan/naproxen sodium group and the placebo group (Table 2).
Satisfaction
PPMQ-R scores reflected greater satisfaction with sumatriptan/naproxen sodium than placebo or previous therapy (pre-study) for the total score (p < 0.001) as well as each subscale score including efficacy, functionality, ease of use, and bothersomeness of side effects (Table 2). Similarly, on the global satisfaction items, significantly greater proportions of patients were satisfied or very satisfied with sumatriptan/naproxen sodium compared with placebo or previous therapy (pre-study) for overall efficacy and overall treatment satisfaction (Figure 3). Significantly greater proportions of patients were satisfied or very satisfied with sumatriptan/naproxen sodium compared with previous therapy (pre-study) for side effects, but not compared with placebo (Figure 3).
Percentage of patients reporting that they were satisfied/very satisfied with sumatriptan/naproxen sodium, placebo, and previous (pre-study) medications.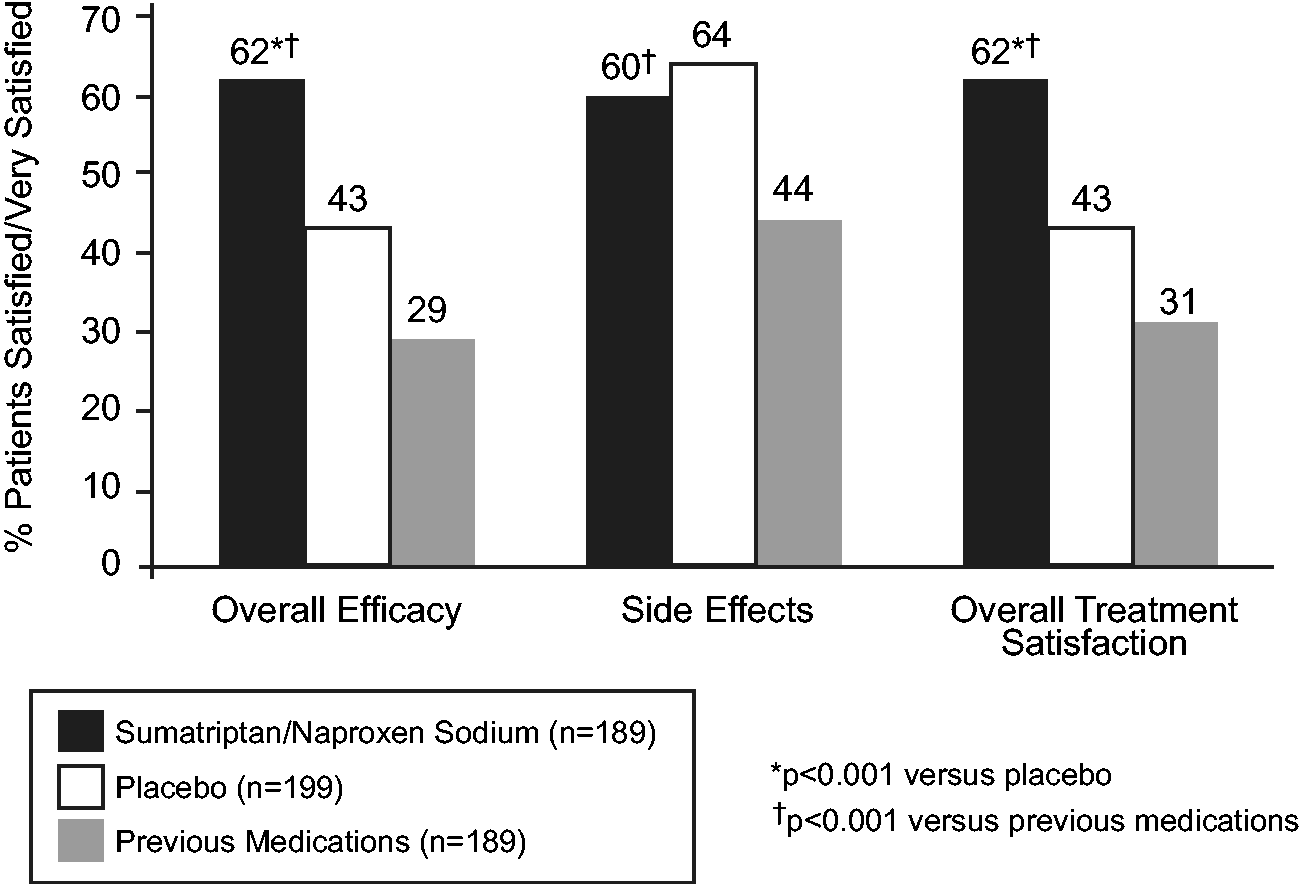
Safety/tolerability
All adverse events.

Discussion
Probable migraine without aura is common among patients consulting for headache and an important variant of migraine in neurologic practice (8–11). The current study shows that sumatriptan/naproxen is more effective than placebo for the acute treatment of probable migraine. Significant differences in favor of sumatriptan/naproxen sodium were observed for the co-primary endpoints of two-hour pain-free response and two- to 24-hour sustained pain-free response and several secondary endpoints. The magnitude of the difference between active treatment and placebo in postdose pain (two-hour pain-free, 29% vs 11%; two- to 24-hour sustained pain-free, 24% vs 9%) in probable migraine was similar to that observed in studies of sumatriptan/naproxen sodium in strict migraine (14).
The satisfaction and functional ability (disability) data support the efficacy data in demonstrating therapeutic benefit of sumatriptan/naproxen sodium over placebo. Patients were more satisfied with sumatriptan/naproxen sodium compared with both placebo and their previous (pre-study) therapy, which for most patients was an NSAID. The magnitude of the differences between active treatment and placebo or previous therapy on the PPMQ-R was statistically as well as clinically meaningful (as it equaled or exceeded the minimal clinically important difference of five points) for the efficacy, functionality, and ease of use subscales as well as the total score. The minimal clinically important difference has not been identified for the tolerability subscale.
Although sumatriptan/naproxen sodium was more effective than placebo for two-hour and two- to 24-hour sustained pain-free responses, it was not more effective than placebo for two-hour pain relief, historically a common primary endpoint in migraine studies (26,27). The latter finding is expected given the lower sensitivity and greater subjectivity of pain relief compared with pain-free response. Pain-free response is a much more rigorous endpoint than pain relief, an endpoint that allows for the presence of residual pain. The high placebo response rate for two-hour pain relief (51%) and the lower one for the more stringent endpoint of two- to four-hour pain relief (42%) support these considerations.
The ICHD-II diagnostic criteria define a probable migraine attack as one that has all but one of the diagnostic features of migraine. As eligibility criteria for the current study required that patients typically have moderate or severe headache pain and headache duration of four to 72 hours (migraine features) but did not require the presence of migraine-associated symptoms, it was expected that patients would often qualify as having probable migraine by virtue of the absence of migraine-associated symptoms. In fact, a low baseline incidence of associated symptoms was reported at the time of dosing in this study compared with representative incidences in patients meeting diagnostic criteria for strict migraine (nausea 5%, photophobia 23%, phonophobia 26% in this study vs nausea 41% to 56%, photophobia 79% to 83%, phonophobia 74% to 83% in a recent study of migraineurs (14)).
Results of this study confirm previous data suggesting that probable migraine episodes are associated with functional impairment (3,6). At screening, mean scores on the HIT-6, which measures the impact of headaches on ability to function at work, school, home, or in social situations, exceeded 60 in both treatment groups, a result reflecting a very severe impact on functional ability. Furthermore, nine of 10 patients in this study indicated some degree of impairment in functional ability before dosing with study medication. Sumatriptan/naproxen sodium compared with placebo significantly improved functional ability on the CDQ, a measure that has been used in numerous migraine studies (21–23).
The adverse-event profile of sumatriptan/naproxen sodium during treatment of a single probable migraine attack in this study was similar to that in previously reported migraine trials (14,16–19). It seems likely that the safety/tolerability considerations that apply to use of sumatriptan/naproxen sodium in strict migraine also apply in probable migraine. Specifically, sumatriptan/naproxen sodium should be prescribed with knowledge of its cardiovascular, cerebrovascular, and gastrointestinal risks in some patients.
The authors would like to acknowledge the strengths and limitations of the study. This is the first study evaluating the acute treatment of probable migraine that has demonstrated efficacy in this population. The study was designed to address previous limitations in the study conducted by Tepper et al. (11), which appears to have contributed to the overall success of this study. Some of the limitations addressed in this study included the following: 1) the use of an e-diary to maximize the number of treated “probable” migraine headaches vs “tension-type” or “migraine,” 2) the prospective inclusion of the intermediate sustained endpoints (one to two and two to four hours), and 3) the use of complete freedom from pain instead of pain relief as one of the two co-primary endpoints. The introduction of the e-diary also has some limitations in terms of generalizability to the patient population, unless patients with probable migraine are using electronic devices to determine which headache to treat with which medication.
Another limitation of the study was the inability to describe the headaches that were not treated. These descriptions may have offered interesting clinical and diagnostic findings, which may have furthered our understanding of the spectrum of headaches and migraine. Lastly, the number of patients who treated with study medication during the study (ITT population) vs those who were eligible to treat (randomized population) was inconsistent with the authors’ understanding of the disability of probable migraine. For example, if probable migraine is disabling, why did not more patients treat with study medication? The following factors may have affected patients’ willingness to treat their probable migraine over the minimum three months allowed: 1) the patient did not recognize probable migraine, 2) the patient’s probable migraine was not triggered over the three months in the study, 3) the patient was not comfortable treating probable migraine with an experimental medicine, 4) the patient did not have his or her study medication, and/or 5) the patient did not have his or her e-diary. The authors acknowledge that this discrepancy cannot be fully explained within this dataset. With limited research in this population compared to migraine, these findings warrant further investigation.
The authors would also like to comment on our use of “ITT population,” which, in this study, is closer to a “complete case analysis” definition since for inclusion, patients were required to treat a headache and have at least one post-baseline assessment vs the traditional definition which is any patient randomized. Applying the traditional definition of ITT population to this study, i.e. a nonprophylactic indication such as the acute treatment of probable migraine where the patient waits for an event before it is treated, would result in significant missing outcome data. In 2010, CONSORT guidelines recommend the use of “complete case analysis” in this situation; however, because the study was designed and conducted in collaboration with the US Food and Drug Administration (FDA) prior to these guidelines, the authors retained the original nomenclature.
In conclusion, sumatriptan/naproxen sodium, a combination therapy targeting both vascular and inflammatory processes in migraine, is the first migraine-specific medication to demonstrate efficacy, improvement in functional ability, and tolerability in prospective, placebo-controlled research in probable migraine without aura. Sumatriptan/naproxen sodium has proven efficacy in probable migraine, a subtype of migraine with similar pathophysiology and disability as migraine but to date without targeted medications to address this unmet medical need. This study establishes a model for randomized trials in probable migraine and may help inform future research on this neglected group of migraine sufferers.
Clinical implications
Sumatriptan/naproxen sodium is the first migraine-specific medication to demonstrate efficacy, improvement in functional ability, and tolerability in placebo-controlled research in probable migraine without aura. This study establishes a model for randomized trials in probable migraine and may help inform future research on this neglected group of migraine sufferers.
Footnotes
Funding
This work was supported by GlaxoSmithKline.
Acknowledgments
The authors acknowledge David Goodman for study management and Shashidhar Kori for contributing to protocol development. The authors thank Kim Poinsett-Holmes, PharmD, (Poinsett Publications Inc) for editorial assistance in preparation of the manuscript.
Conflicts of interest
This study was sponsored and conducted by GlaxoSmithKline. SS, JG, SA, and RBL have consulted for or conducted research funded by GlaxoSmithKline and other pharmaceutical companies. SAM, SEL, JW are employees of GlaxoSmithKline. FD, MCR were employees of GlaxoSmithKline at the time this study was conducted and reported. The work of JS, a professional medical writer, on this manuscript was funded by GlaxoSmithKline.