
Abstract
This review outlines the pearls and pitfalls of calcitonin-gene related protein (CGRP) immunohistochemistry of the brain.
Pearls
In 1985, CGRP was first described in cerebral arteries using immunohistochemistry. Since then, cerebral CGRP (and, using novel antibodies, its receptor components) has been widely scrutinized. Here, we describe the distribution of cerebral CGRP and pay special attention to the surprising reliability of results over time.
Pitfalls
Pitfalls might include a fixation procedure, antibody clone and dilution, and interpretation of results. Standardization of staining protocols and true quantitative methods are lacking. The use of computerized image analysis has led us to believe that our examination is objective. However, in the steps of performing such an analysis, we make subjective choices. By pointing out these pitfalls, we aim to further improve immunohistochemical quality.
Recommendations
Having a clear picture of the tissue/cell morphology is a necessity. A primary morphological evaluation with, for example, hematoxylin-eosin, helps to ensure that small changes are not missed and that background and artifactual changes, which may include vacuoles, pigments, and dark neurons, are not over-interpreted as compound-related changes. The antigen-antibody reaction appears simple and clear in theory, but many steps might go wrong. Remember that methods including the antigen-antibody complex rely on handling/fixation of tissues or cells, antibody shipping/storing issues, antibody titration, temperature/duration of antibody incubation, visualization of the antibody and interpretation of the results. Optimize staining protocols to the material you are using.
Introduction
Immunohistochemistry, a morphology-based technique first described in the 1930s (1) and that became brisk during the 1960s (2**), provides an important and valuable tool in many fields of research. The method is based on the localization of an antigen, usually proteins, in tissue sections and cells by the use of an antibody that specifically binds to the antigen. The antigen-antibody complex is subsequently visualized by a marker, such as a fluorescent dye, that has the disadvantage of fading over time, or the widely used and stable horseradish peroxidase-diaminobenzidine (DAB). The achieved signal should be distinct and at the same time retain the morphology.
Thanks to developments in molecular biology, it is possible to synthesize proteins of interest and to raise antibodies to these proteins. In addition, sensitivity of antibodies and detection systems has rapidly increased (3). Advancements over the years within immunohistochemistry have resulted in an enormous number of scientific publications. Searching for “immunohistochemistry” on PubMed produces approximately 517,000 hits; searching for “Western blot,” another method using the antigen-antibody complex, gets approximately 192,000 hits. These counts demonstrate the usability of the method, but also the applicability of the antigen-antibody complex in science. The counts also suggest that several pitfalls might exist that can lead to misinterpreted results.
Migraine is a disorder affecting more than 10% of the general population. Currently, migraine is considered a neurovascular disorder involving activation of the trigemino-vascular system with disturbance in the brainstem/brain, demonstrated with positron-emission tomography (PET) (4). Intense research has been carried out to identify signal molecules associated with the trigeminal system. The only neuronal messenger so far reliably demonstrated in migraine attacks is the neuropeptide calcitonin gene-related peptide (CGRP) (5–7).
CGRP in neural tissue was first described in 1983, where it was shown that the calcitonin gene results in the production of a messenger RNA, generating CGRP, in neural tissue distinct from that in thyroidal 'C' cells (8**). The discovery of CGRP in neural tissue consequently led to intense scrutiny of the CGRP distribution. In 1985, Edvinsson and colleagues (9**) described CGRP distribution of perivascular nerve fibers and in the trigeminal ganglia, and later on CGRP fibers in non-vascular parts of the dura (10). In addition, early immunohistochemical studies demonstrated CGRP in the cerebellum (11). Since then, the immunohistochemical technique has widely been used to further investigate the distribution of CGRP in the brain, in particular the neurovascular and trigeminovascular systems suggested to be involved in migraine (12–17).
Advances in understanding CGRP suggest that the pathophysiology of migraine is far more complex than was originally thought, and that vascular activation may be just one of many factors involved. Although CGRP has a number of effects, its most pronounced actions are intracranial vasodilatation and transmission of nociception (18). There is a correlation between CGRP release and pain in migraine (19,20) that points toward the potential usefulness of a specific antagonist in the treatment (21). The CGRP receptor has long been regarded as an important target for the development of antimigraine therapies. Telcagepant is an orally available CGRP-receptor antagonist and, together with BI 44370 TA (22), have shown efficacy in late stages of clinical testing for the treatment of acute migraine (23,24). It remains to be determined if the antimigraine action of CGRP-receptor antagonists is mediated via central or peripheral mechanisms, or both (25). The site of action of the CGRP receptor antagonists is debated, but presently both the peripheral and central ends of the trigeminovascular system are considered likely targets (26).
This review outlines the pearls and pitfalls of CGRP immunohistochemistry of the brain. We pay special attention to the surprising reliability of results over time. However, since negative results are rarely published and questionable methodology or inexperience might go unnoticed, we focus on pitfalls with the expectation that the advice given in terms of fixations, antibody clone and dilution, and interpretations, for example, might assist scientists and further improve immunohistochemical quality.
Pearls
In 1985, Edvinsson (27) first showed that cerebral arteries in the circle of Willis were surrounded by a dense network of CGRP fibers, whereas small pial arteries on the cerebral convexity usually were accompanied by only a few fibers (9**) (Figure 1). Furthermore, blood vessels in the dura contained a moderate number of CGRP fibers, which were also found in non-vascular parts of the dura (13). CGRP antiserum (code no. 8427, Milab, Malmö, Sweden) was at that time raised in rabbit against synthetic albumin-conjugated rat CGRP (Peninsula, Belmont, CA, USA). In 2012, we can still show the same distribution pattern of CGRP using immunohistochemistry and commercially available antibodies (monoclonal CGRP, ab81887, host mouse, 1:100, Abcam, UK) (Figure 2).
Cat pial artery immunostained with calcitonin gene-related protein CGRP (9**) (published with permission). Circle of Willis. (a) cat (13) (with permission), (b and c): rat (Warfvinge, 2012, unpublished observations).

The detailed examination of CGRP receptor components has been made possible with the recent development of a series of specific antibodies against calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein type 1 (RAMP1) (28**). This is of particular interest in understanding how the recent CGRP receptor antagonists exert their antimigraine effects.
Immunohistochemical studies performed on the trigeminal system have shown CGRP immunoreactivity in about 50% of the neurons of human and rat trigeminal ganglion (10,17,29). Edvinsson and colleagues (28**) demonstrated the detailed distribution and quantification of CLR and RAMP1 in human trigeminal ganglion. The CGRP receptor is localized to neurons (mainly large) as well as satellite glial cells (surrounding neurons), while CGRP is expressed only in the neurons of small to medium size and these lack CGRP receptor components (Figure 3). This suggests that if CGRP is released within the ganglion, then intraganglionic CGRP may act on satellite glial cells and on large-sized neurons.
Upper panel. Cat trigeminal ganglion immunostained with calcitonin gene-related protein (CGRP) (9**) (with permission). Lower panel. Distribution of CGRP, calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP1) in human and rat trigeminal ganglia (28**) (with permission).
The trigeminal ganglion, storing CGRP and its receptor components, projects peripherally to the intracranial vasculature and dura mater and centrally to regions in the brainstem with Aδ- and C-fibers. This constitutes an essential part of the pain pathways activated in migraine attacks. Therefore it is of importance to identify the regions within the brainstem that process nociceptive information from the trigeminovascular system, such as the spinal trigeminal nucleus (STN) and the C1-level of the spinal cord. Liu and colleagues (30–33) showed projections of thin fibers marked with wheat germ agglutinin-conjugated horseradish peroxidase (WGA-HRP) to laminae I/II and thick fibers labeled with cholera toxin subunit b (Ctb) to laminae IV/V emanating in cranial vessels of various locations (25).
Immunohistochemistry was used to study the distribution and relation between CGRP and its receptor components CLR and RAMP1 in human and rat STN at the C1-level (16). Fibers expressing CGRP and its receptor components occur in STN (Figure 4) and C1, however, in laminae I/II only. This was a bit unexpected because the tracing studies predicted the A-fibers to project to laminae IV/V rather than I/II (31). Differences in the CGRP expression between the species were observed. In addition the distribution was somatotopically organized to different parts of the trigeminal nucleus caudalis (TNC)/C1 regions. It was also demonstrated that fibers and neurons expressed CGRP close to the central canal, which suggests that CGRP may have a function within this area. With confocal microscopy the detailed relation between CGRP- and CLR/RAMP1-containing fibers revealed that CGRP and the receptor were always detected in different fibers (16). This suggests that they communicate via synapses. In addition, presynaptic action (within the same nerve fibers), which has been suggested before (17), is disputed since there was no co-localization between CGRP and the receptor components.
A. Calcitonin gene-related protein (CGRP) immunoreactivity in rat subthalamic nucleus (STN). B. CGRP-positive neurons in the region of the inferior olive. Thanks to Dr S. Eftekhari. For further information about distribution, see Eftekhari and Edvinsson (16).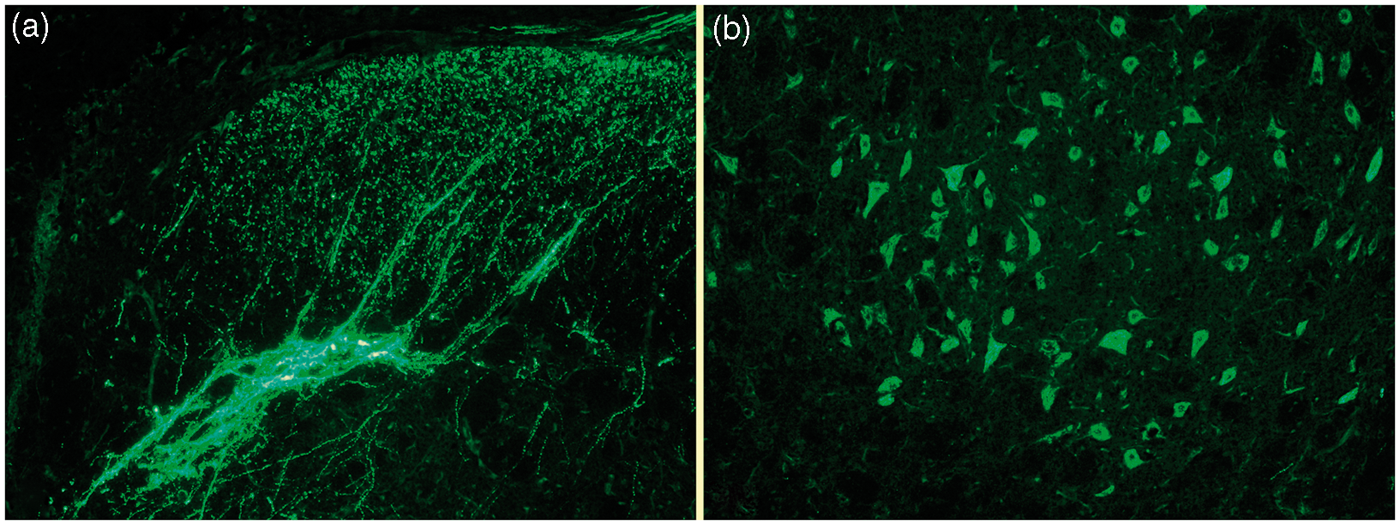
We examined the distribution of CGRP and its receptor in the cerebellum (34). Early immunohistochemical studies have demonstrated CGRP in the cerebellum (11). The CGRP immunoreactivity was localized in the Purkinje cell and in its elaborated dendrite tree. Our observations support these findings and, furthermore, demonstrate that there is a rich expression of CGRP and CGRP receptor components in the cerebellum. We could demonstrate that CGRP was found in the cytoplasm of the Purkinje cells and the receptor components in the Purkinje cell membrane, both in the main cell body and in the dendrites (Figure 5). The findings point toward a functional role of CGRP in cerebellar Purkinje cells. The early PET studies on migraine attacks showed activation not only in the brainstem but also in the cerebellum (4). Recent advances in the biology of the cerebellum indicate that there may be a role in nociception. Hence, a target of the CGRP receptor antagonists, which have demonstrated improvement in migraine pain and associated symptoms, could be cerebellar CGRP receptors.
Rat cerebellar Purkinje cells.
There are some functional data that suggest an interaction between the trigeminal and sphenopalatine ganglia (SPG). Cluster headache is associated with activation of both ganglia since there is co-release of CGRP and vasoactive intestinal peptide (VIP) (35). Treatment with sumatriptan aborts both symptoms of parasympathetic activation and neuropeptide release, presumably by the triptan acting as inhibitor on the sensory nervous system via a presynaptic mechanism or through modifying the formation of CGRP (36).
We investigated the distribution of CGRP and its receptor in human and rat SPG. CGRP immunoreactive fibers were frequently found intraganglionic in the SPG in the vicinity of neurons (37). CLR immunoreactivity was observed in satellite glial cells (SGCs) as well as in nerve fibers, but not in SPG neurons. RAMP1 immunoreactivity was, however, localized in many SPG neurons and SGCs (Figure 6). Thus, the two CGRP receptor components together were found in the SGCs. Our results suggest a possible direct sensory influence in the parasympathetic cranial ganglia. The sensory CGRP-containing fibers probably originate in the trigeminal ganglion, project to the SPG and act on CGRP receptors on SPG neurons and SGCs.
Receptor-activity modifying protein 1 (RAMP1) immunoreactivity of rat sphenopalatine ganglion (SPG). Thanks to Dr A. Csati. For further information, see Csati et al. (37).
Migraine is considered to involve the activation of sensory trigeminal pain neurons that supply intracranial blood vessels and the dura mater. It is suggested that local activation of these sensory nerves may involve dural mast cells as one factor in local inflammation causing sensitization of meningeal nociceptors. Whatever the cause of activation of the peripheral nociceptors in the intracranial vasculature and meninges, the input that results in headache is abnormal (peripheral sensitization). However, the detailed distribution and neurotransmitter content of the sensory A- and C-fibers and their relations to dural mast cells are unclear. We therefore studied the distribution of CGRP, and CLR and RAMP1 in whole-mounted rat dura mater (Eftekhari et al. (38)). CGRP is expressed in thin un-myelinated fibers, suggesting C-fibers (Figure 7). CLR and RAMP1 are instead expressed in the thicker myelinated fibers, suggesting A-fibers. This supports the view of CGRP being expressed in C- fibers, which may release the peptide to act post-junctionally on A-fibers and mast cells that also express CLR and RAMP1.
Whole mount rat dura mater. Calcitonin gene-related protein (CGRP) is expressed in thin un-myelinated fibers, suggesting C-fibers. Thanks to Drs S. Eftekhari and A. Blixt.
To conclude, much of the results reported on brain CGRP in the 1980s and 1990s are still valid, even though the opportunity to present the results in a reader-friendly way has improved tremendously.
Pitfalls
Technical pitfalls
Fixation
The purpose of fixation is to prevent degradation and to stabilize structures within tissues or cells. Accordingly, it is important that tissues or cells have access to a fixative as rapidly as possible. An inappropriate fixative may destroy the binding site on the antigen that the antibody recognizes. The fixation procedure varies from laboratory to laboratory and there have been numerous attempts to standardize the procedure, especially in clinical work (39). Nevertheless, standardization remains a great challenge mainly because of variable conditions, such as fixative used.
Formalin has over the years remained the universal fixative for routine histology and immunohistochemistry. Consequently, antibodies used for diagnostics have been developed to detect epitopes in their formalin-fixed form. Non-formalin fixed and/or alternative fixation methodologies are discouraged in clinical work largely because performance data are limited and extrapolation from formalin fixed is unreliable. For similar reasons, one might argue that formalin is the preferential fixative also in the research laboratory. However, a few immunohistochemical protocols request snap freezing and/or acetone fixation, for example. Cryopreservation is a nonchemical preservation where the proteins are presented in a less altered way compared to native conformation of the protein. However, the morphology is often compromised by the freezing. In addition, acetone fixation often results in shrinkage of the tissue. Consequently, the type of fixation must be decided according to data from the distributor, either commercially available data or single laboratory data. Generally, however, a tissue fixed in formalin-based fixatives shows a higher level of immunoreactivity and provides superior morphology (40).
Formalin fixation differs between laboratories regarding duration, concentration, pH, buffers and temperature. All these parameters strongly affect immunostaining. When it comes to the duration of fixation, it is likely that certain antigens have optimal durations of fixation. It has been shown that only short fixation time reveals estrogen receptor (41) and that progressive fixation displays a time-related loss of antigens (42). In a recent study, it was shown that fixation artifacts were present in approximately 30% of more than 1700 magnetic resonance images of perfused mouse brains (43). The authors emphasized the importance of correct perfusion flow rate, and the form and concentration of perfusate.
For preparation of brain tissue, transcardial perfusion is preferred over immersion fixation because it utilizes the vascular network to deliver fixative rapidly and evenly to the brain tissue. The cerebral vasculature is highly arborized and all cells are, on average, within 100 µm of a vessel (44).
Morphological evaluation
It is important to evaluate the fixation of the tissues or cells by routine histology, for example, hematoxylin-eosin (Htx-eosin). It is a fast and relatively inexpensive way to investigate the morphology, which will give an indication about the quality of the fixation but also, for example, depict infarction areas, although one should be aware of the limitation in verification of early cell death. For this, other methods (45) are recommended such as FluorJade, Caspase3 immunohistochemistry or TdT-mediated dUTP nick end labeling (TUNEL) staining (Figure 8). Primary morphological evaluation helps to ensure that small changes are not missed and that background and artifactual changes, which may include vacuoles, pigments, and dark neurons, are not over-interpreted as compound-related changes.
Detection of apoptotic cells using TdT-mediated dUTP nick end labeling (TUNEL). Arrows point at hippocampal positive cells and arrow heads at negative cells. Lower panel. Higher magnification of hippocampal cells. Green: TUNEL; red: tetramethyl rhodamine isothiocyanate (TRITC) filter; blue: 4′-6-diamidino-2-phenylindole (DAPI) nuclei staining. Thanks to Dr S. Johansson.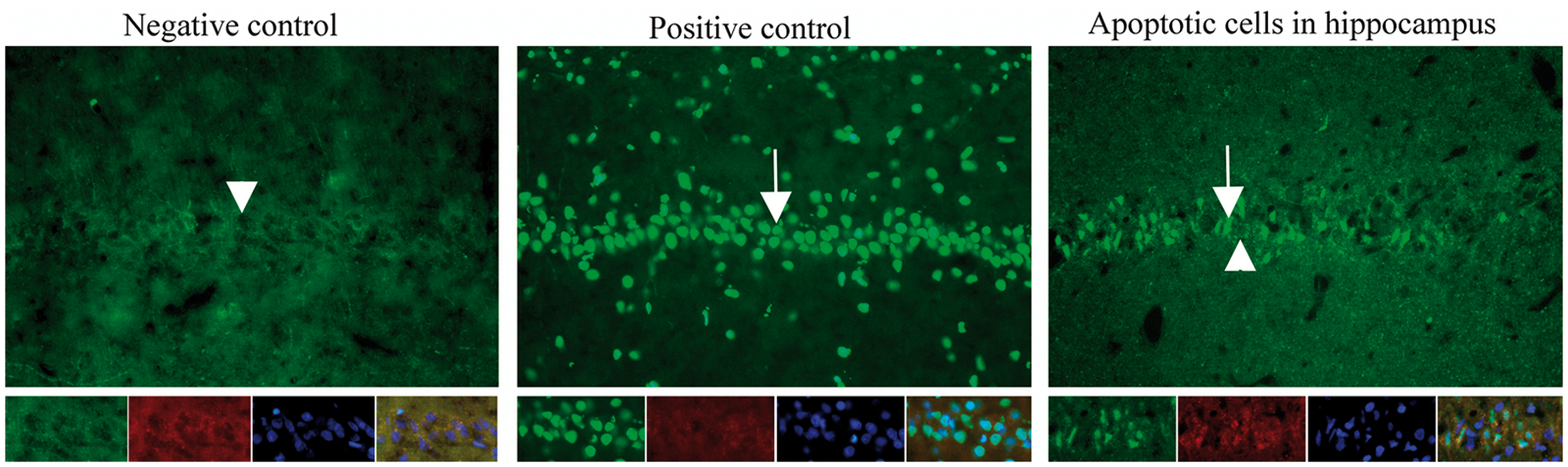
Antigen-antibody reaction
The antigen-antibody reaction must be validated (46). A validation process demonstrates that the performance characteristics of the method are suitable for its intended use. For antibodies, this means that they should be specific, selective and reproducible in the context for which they are used.
The correct use of controls includes the staining of a negative control, which is a cell line or tissue that is known not to express the protein of interest (preferably knockout animals), and a positive tissue that over-expresses the protein. It should be noted, however, that the examined specimen, and the negative and positive controls, have most likely not been handled in the same manner, e.g. fixation procedures (concentration, temperature, duration – variables that have an impact on the immunohistochemistry), which suggests that the use, whenever possible, of internal controls is optimal.
The use of heat-induced antigen-retrieval protocols permits retrieval of a vast range of specific proteins from formalin-paraffin sections (47). The method, although highly variable between laboratories, greatly enhances the antigenicity and reproducibility.
Western blot analyses should be performed to confirm that the size of the detected protein is correct and the antibody is specific. The first indication that the antibody is specific for the selected target would be a single band at the known molecular weight. In Western blots, proteins are transferred onto a protein-absorbing membrane that then is incubated with the antibody against the desired protein. The control should contain equal protein loading. This is usually performed with an antibody that recognizes a protein with relatively constant expression. ß-actin and glyceraldehydes 3-phosphate dehydrogenase (GAPDH) are often used as a loading control. However, it has been shown that ß-actin (48) and GAPDH (49) are not reliable loading controls in Western blots. It is recommended that different dilutions of a positive control be run in the same gel as the sample.
To control the specificity of the antibody, a blocking peptide could be used. It does not prove selectivity of the antibody since off-target binding activity of the antibody will also be inhibited by pre-absorption with the blocking peptide. So while blocking peptides can prove that an antibody is bad, when non-specific staining is seen in the presence of the peptide, they cannot prove that an antibody is good. In addition, titration experiments are necessary to optimize the staining for each antibody. To demonstrate reproducibility, different lots of antibodies on different days should be used, and a new antibody should be compared to a previously validated antibody. Issues during shipping, storage and antibody contamination are of course of great importance in achieving reliable results.
Digital image mismatches
The use of computerized image analysis has led us to believe that our examination is objective. However, in the steps of performing such an analysis, we make choices. Tadrous (50) concludes that a great variety of personal choices, based on opinion and judgment, is required to construct any given image analysis method. The computer performs the analytical process, but a human being decides on numerous influences in the design such as tissue preparation, image capture (including selecting the area for capture), optional pre-processing (e.g. color balance), image segmentation (region of interest (ROI)), optional post-processing, calculation and interpretation. This has been discussed in detail by Tadrous (50) and Leong (3).
Both clinical diagnostics and laboratory science have a need to quantitate the stain reaction. Currently, quantitation most often involves measurement of intensity and extent of the stain, without considering that, for example, the thickness of the section has an impact on the nuclei intensity (51). The practice of quantifying immunohistochemical stainings is, at best, approximate and only semi-quantitative. Thus, the development of true quantitative methods is crucial.
Sustainable stereological methods were developed during the 1980s and 1990s by Gundersen and West (52). They designed unbiased stereological counting methods that offered a tool to obtain absolute cell numbers in a three-dimensional structure from a two-dimensional tissue slide where the computer determines the placement of random counting frames and the depth (z-axis) was equal to the section thickness. These methods do not take the subjectivity of intensity into consideration. However, randomized counting areas are determined.
Superimposition of photographed images from different labeled antigens and color manipulation can produce clear distinction between two or more colors (Figure 9). If double-labeling is performed, sometimes better staining patterns are observed when primary and secondary antibodies are added sequentially.
Rat retinal “rosette” formation. Green: embryological green fluorescent protein (GFP) transduced cells grafted to the subretinal space of pigs; red and blue: sections from the grafted eye immunostained with antibodies against retinal rod and cone proteins. Merged image demonstrates grafted cells that have matured to cells containing either rod or cone proteins.
Our methods
Fixation
Formalin immersion fixation is not ideal because of, in part, a slowing down penetration process. This partly slow process can best be described as the penetration-fixation paradox (53). This means that if a block of tissue is fixed in formalin, cells of extreme dimensions of the block will have different morphological properties compared to that of cells a millimeter or so further within the block. However, a well-dissected specimen should overcome the problem.
Four percent paraformaldehyde in phosphate-buffered saline (PBS, pH 7.2) may penetrate a thin tissue within a few hours. However, formaldehyde penetrates only 2.4 mm in 24 hours, leaving a well-fixed outer perimeter and a poorly fixed inner core in 10 mm-thick specimens (54). This means that the dura mater, for example, needs only one to two hours of fixation to display a well-preserved tissue (Eftekhari et al. (38)) and the retina (55) or well-dissected vessels (56), only two to four hours of fixation. It also means that in a context of formalin-working antibodies, the morphology will be adequate in thin tissues like the retina. On the other hand, the brain or pieces of the brain need far longer fixation. Because of the slowing down of the penetration, the deeper/larger the tissue, the poorer fixation demonstrates the penetration-fixation paradox. Perfusion combined with post-fixation will be the more appropriate method of fixation of the brain.
Immersion fixation is followed by wash in Sörensen’s phosphate buffer with rising concentrations of sucrose. Normally 10% sucrose (until the specimen sinks showing total sucrose immersion of the specimen) followed by 25% (until the specimen sinks) gives appropriate cryo-protection. Prior to sectioning, the specimen is embedded (on dry ice and 70% alcohol) in a gelatin medium (30% egg albumin and 3% gelatin in distilled water) and cryo-sectioned at 10 µm. The sections are stored at −20°C until use.
Transcardial perfusion normally starts with a prewash of PBS for 2 minutes at a rate of 50 ml/min then is followed by 4% paraformaldehyde in PBS for 5 minutes. However, a lower perfusion rate plays a role in minimizing fixation artefacts (40). The temperature of the fixation liquids should be kept at room temperature, according to Paavilainen et al. (40) For cryostat sectioning and good morphology, post-fixation in formalin one to three hours and thereafter immersion in Sörensen’s phosphate buffer with sucrose, as described above, are performed.
Htx-eosin
To have a clear picture of the tissue/cell morphology, it is a necessity to perform Htx-eosin (or a similar morphological staining) prior to immunohistochemistry. Prior to staining, the sections are allowed to dry for 10–15 minutes at room temperature. For staining, a standard protocol is used (Htx 4 min, water rinse, eosin 30 sec–1 min).
Immunohistochemistry
For immunohistochemistry, sections are thawed and washed for 10 min in PBS pH 7.2 containing 0.25% Triton X-100 (PBST). The sections are then incubated overnight at +4°C with primary antibodies diluted in PBST containing 1% bovine serum albumin (BSA) and 3% normal serum. After incubation with primary antibodies, sections are equilibrated to room temperature, rinsed in PBST for 3 × 15 min and exposed to secondary antibodies in PBST and 1% BSA for 1 hour at room temperature in a dark room. The sections are subsequently washed with PBST for 3 × 15 min. Vectashield anti-fading medium containing the nucleus-staining 4′-6-diamidino-2-phenylindole (DAPI) (Vectashield, Vector Laboratories, Burlingame CA, USA) is used as mounting media. Omission of the primary antibody serves as a negative control for all antibodies. All antibodies are applied in at least three independent staining sessions in order to validate reproducibility.
Microscopic analysis
Sections are examined and images obtained using a light and epifluorescence microscope (Nikon 80i, Tokyo, Japan) coupled to a Nikon DS-2MV camera. Image analyses are conducted using NIS basic research software (Nikon, Japan). Adobe Photoshop CS3 (v.8.0, Adobe Systems, Mountain View, CA, USA) is used to visualize co-labeling by superimposing the digital images, and are processed for brightness and contrast.
Recommendations
Having a clear picture of the tissue/cell morphology is a necessity. A primary morphological evaluation with Htx-eosin, for example, helps to ensure that small changes are not missed and that background and artifactual changes, which may include vacuoles, pigments, and dark neurons, are not over-interpreted as compound-related changes.
The antigen-antibody reaction appears simple and clear in theory, but many steps might go wrong. Remember that methods including the antigen-antibody complex rely on handling/fixation of tissues or cells, antibody shipping/storing issues, antibody titration (often more than 1:1000), temperature/duration of antibody incubation, visualization of the antibody and interpretation of the results. Until standardization in immunohistochemical processing is achieved, optimize staining protocols to the material you are using (3).
Footnotes
Acknowledgements
Thanks to Drs Sara Johansson, Sajedeh Eftekhari, Frank W. Blixt and Anett Csati, and the Lundbeck Foundation, DK.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.