
Abstract
Introduction: Familial hemiplegic migraine (FHM) is characterized by the familial occurrence of migraine attacks with fully reversible transient hemiplegia. Mutations in three different genes have been identified; CACNA1A (FHM1), ATP1A2 (FHM2) and SCN1A (FHM3). Besides hemiplegia, several other symptoms have been described in FHM 1–3 mutation carriers, including epilepsy and cerebellar symptoms.
Case report: We describe two patients in whom hemiplegic attacks were not the presenting symptom, but in whom an otherwise unexplained head tremor led us to search for FHM mutations. Both patients carried a mutation in the CACNA1A gene.
Discussion: CACNA1A mutations can give significant symptoms other than (hemiplegic) migraine as reason for presentation.
Introduction
Hemiplegic migraine (HM) is a rare migraine subtype characterized by transient hemiplegia during the aura phase of the attacks (1). Hemiplegic migraine may run in families (familial hemiplegic migraine; FHM), but a sporadic form (sporadic hemiplegic migraine; SHM), which is clinically indistinguishable from FHM, also exists (1–5). FHM has an autosomal dominant mode of inheritance and is a genetically heterogeneous condition with mutations in at least three different genes: CACNA1A (FHM1), ATP1A2 (FHM2) and SCN1A (FHM3) (6–8).
HM is the only migraine disorder for which multiple pathogenic genetic mutations have been found so far (2–5,9,10). Currently, many FHM mutations are known, associated with a broad spectrum of symptoms, including migraine, epilepsy and cerebellar symptoms (3–5,9–19).
Here, we describe two patients in whom an otherwise unexplained head tremor, but not HM, led us to search for FHM mutations.
Case report
Patient 1
A 30-year-old right-handed woman presented with a permanently present tremor of head and neck that she had had for 12 years. The complaints had increased gradually over the previous years and worsened intermittently during periods of psychic stress. During the previous few months she had suffered from intermittent headaches irradiating from the neck, mainly during periods of stress. The headache was not accompanied by phonophobia, photophobia, nausea or vomiting. Two years earlier she had had an attack that started with tingling of the right foot, which gradually went up to the upper leg and arm. Fifteen minutes later the lower right side of her face was hanging down. Her speech had been unintelligible. Later she experienced dark spots in both visual fields. The symptoms lasted half an hour and were not followed by headache.
One year earlier she had another episode of tingling of the foot irradiating to the leg, but without other symptoms. Her medical history was unremarkable otherwise.
Her father and sister were known to suffer from migraine with visual aura, without hemiplegia. They did not have any kind of tremor. At neurological examination the only abnormality was titubation of the head (‘no-no’), without hand tremor or dystonia. MRI of the brain showed cerebellar atrophy, especially of the superior cerebellar vermis (Figure 1a and 1b). Tremor registration of the splenius and sternocleidomastoid muscles revealed a 3–5 Hz tremor. Direct sequencing of the CACNA1A gene showed that she carried a heterozygous c.2003 A>C substitution in exon 16 (GenBank accession number X99897). This point mutation resulted in an amino acid change from glutamic acid to alanine at position 668 (p.Glu668Ala). The parents of the proband were tested for the mutation and the father (28 years older than the patient) appeared to carry the same mutation as the proband. The sister (2 years younger) was not available for testing. The mutation is located in a strongly conserved functional domain in the extracellular loop between segments 5 and 6 of domain II (IIS5 and IIS6; Figure 2) and was absent in over 300 controls.
(a) Axial brain MRI of patient 1. (b) Sagittal brain MRI of patient 1. Both scans show cerebellar atrophy especially of the superior cerebellar vermis. Schematic representation of the pore-forming α1 subunit of the voltage-gated CaV2.1 calcium channel encoded by the CACNA1A gene. The α1 subunit consists of four homologous transmembrane domains (I–IV). Each of these domains consists of six transmembrane segments. The p.Glu668Ala mutation is located between II Segment 5 and II Segment 6.

Patient 2
A 57-year-old woman suffered from a permanent tremor of the head and hands for 13 years. In 2007 she presented to our outpatient clinic because the tremor had worsened. Her medical history mentioned epilepsy for which she was using valproate (up to 500 mg bid) for at least 15 years (a relationship with the tremor had been excluded in the past by temporarily interrupting its administration), and she had been seizure free for some years. She also suffered from migraine without aura, for which she took propranolol and sumatriptan, but during the past few years she had been attack free. The propranolol nevertheless was continued as she also experienced some relief with respect to the tremor.
Several family members, in at least three generations, suffered from migraine, including her daughter who had migraine with aura. Our patient had ten children, six of whom had already died from unrelated causes.
At neurological examination only a tremor of the head and hands (position tremor) was found. She was only 1.51 m tall. A computed tomography (CT) scan of the brain in 1993 had been normal and EEG’s at that time had shown bilateral epileptic abnormalities (interpreted as primary generalized epilepsy). An MRI of the brain in 2007 showed cerebellar atrophy (Figure 3). Cerebrospinal fluid was unremarkable, with normal lactate and pyruvate. Tremor registration of the splenius and sternocleidomastoid muscles revealed a 3.2–4.9 Hz tremor. Examination of the mitochondrial DNA (in blood) did not reveal abnormalities consistent with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibres (MERRF) or Leigh syndrome/neurogenetic muscle weakness, ataxia, retinitis pigmentosa (NARP). Direct sequencing of the CACNA1A gene revealed a heterozygous c.4109 G>A substitution in exon 26 (Genbank X99897), resulting in an amino acid change from cysteine to tyrosine (p.Cys1370Tyr). This mutation was previously described as C1369Y in a FHM family (11).
Axial brain MRI of patient 2, showing cerebellar atrophy.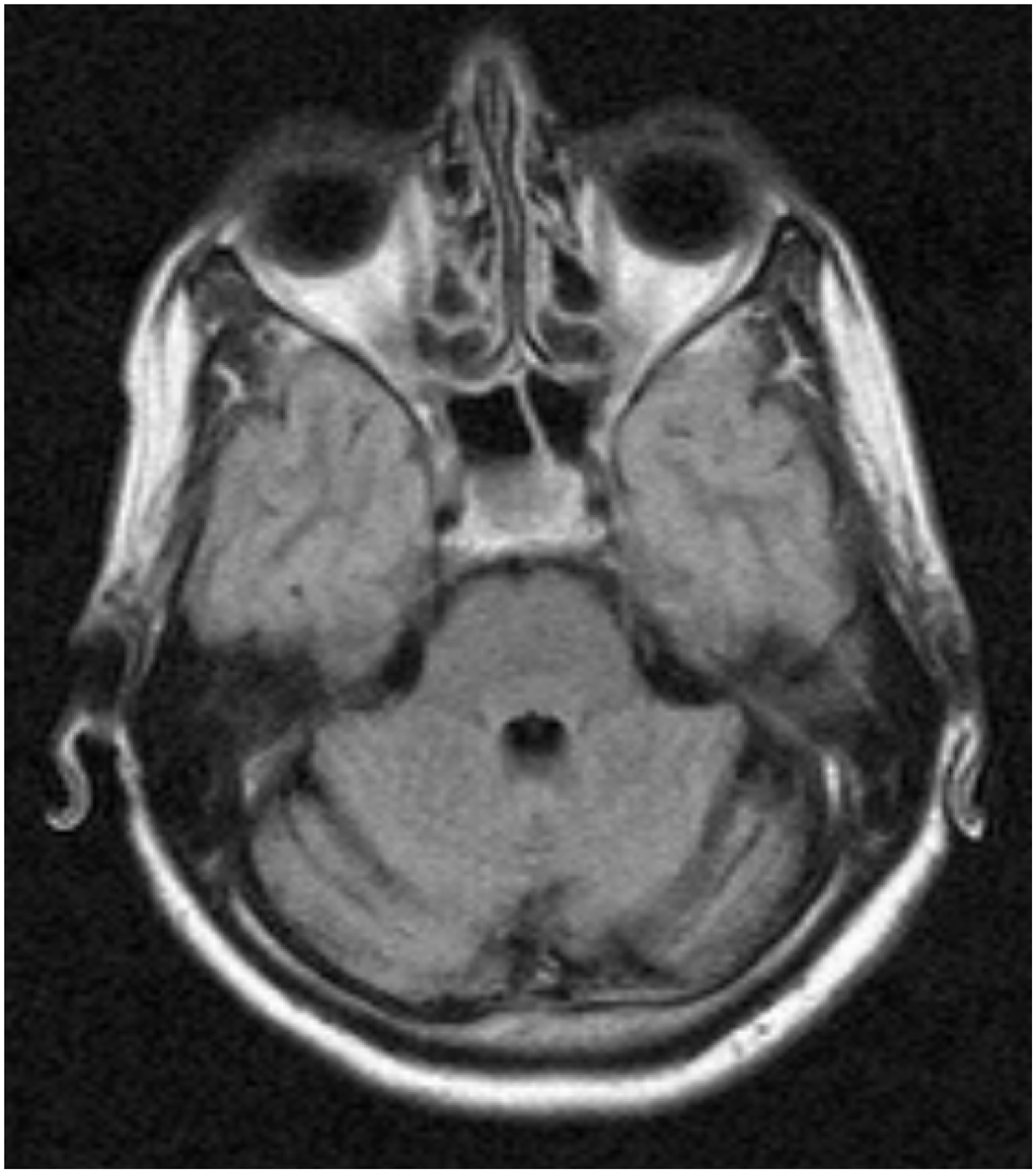
Discussion
We describe two patients with a mutation in the CACNA1A gene, in which the reason for presentation was not HM. In both patients an otherwise unexplained interictal head tremor was the main complaint. Titubation of the head at examination was the reason to order MRI scans, and cerebellar atrophy was found. Both patients underwent tremor registration of the splenius and sternocleidomastoid muscles. The frequency in patient 1 was 3–5 Hz and in patient 2 was 3.2–4.9 Hz. No firm conclusion with respect to the distinction between alternate and synchronous character could be drawn. As the diagnostic validity of quantitative computed tremor analysis with accelerometers has not been established, we did not perform repeat studies of this (20).
Patient 1 suffered from tension type headache (1), a typical migraine aura without headache once or twice, and once a motor aura; however, she did not fulfil the criteria of SHM as the motor signs were restricted to one attack only (and not followed by headache). Patient 2 suffered from migraine without aura and generalized epilepsy. The findings at neurological examination and on MRI, in combination with symptoms of a typical aura (without headache) in patient 1 and epilepsy and migraine without aura in patient 2, made us decide to screen the CACNA1A (FHM1) gene for mutations. Both patients appeared to carry a CACNA1A mutation. Mutation p.Cys1370Tyr was previously published showing a variety of clinical symptoms. One patient had cerebellar ataxia, but it is unknown whether this was also present interictally. A tremor was not described in this patient (11). The p.Glu668Ala mutation is a novel mutation that is located in a highly conserved functional domain of the CACNA1A gene and was absent in 300 healthy controls, indicating pathogenicity.
The CACNA1A gene on chromosome 19p13 encodes the pore-forming α1A-subunit of Cav2.1 P/Q calcium channels which are expressed both pre- and postsynaptically in the central nervous system and neuromuscular junctions, cerebellum (especially in Purkinje cells, which are the source of cerebellar output), cerebral cortex, thalamus and hypothalamus. The channels are responsible for polarization and thus for neuronal function (5,7,9–11,15). Several missense mutations in this gene have been identified, causing a broad spectrum of symptoms.
Mutations in the CACNA1A gene can also cause episodic ataxia type-2 (EA2) (6) and spinocerebellar ataxia type-6 (SCA6) (21). SCA6 is characterized by a late-onset, slowly progressive cerebellar ataxia and atrophy with oculomotor disorders, incoordination and dysarthria, but no epilepsy (4,9,15). Marked cerebellar atrophy may be present on MRI (7). EA2 mutations may cause recurrent or interictal cerebellar symptoms, and epilepsy has been reported (9,15). FHM1 is mainly caused by missense mutations (22), as were identified in our patients.
The most striking characteristic of the patients described here was the titubation of the head in combination with the cerebellar atrophy on MRI. According to previous studies and case reports, cerebellar atrophy on MRI is known to be related to FHM (16). In one population-based study, a patient was described in whom head tremor was one of the symptoms, but it was not clearly described as the most significant symptom (14). Intention tremor has been associated with CACNA1A mutations before: Dichgans et al. (7) reported a patient with a T666M mutation and a patient with a R1347Q mutation with an intention tremor, but no head tremor. Zwingman et al. (23) reported ataxia, unstable gait and an intention tremor in chemically mutagenized male mice, caused by a rkr mutation – an allele of CACNA1A. Therefore, as far as we know, there are no previous reports of HM patients with a tremor of the head and neck muscles (titubation) as the most significant presenting symptom.
According to the IHS criteria, motor aura symptoms are currently obligatory to diagnose HM (1). As penetrance of FHM mutations is incomplete, not all mutation carriers have HM (4,9,12). Therefore a search for FHM mutations could be advised in patients presenting with symptoms that are otherwise unexplained. Here, we add titubation of the head to this list.
Footnotes
Acknowledgements
We thank H.B.M. van Lieshout (Amphia Hospital, Breda, The Netherlands) and M.P.W.A. Houben (Atrium Medical Centre, Heerlen, The Netherlands) for providing the tremor registrations of the patients.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.