
Abstract
Background
Familial hemiplegic migraine (FHM) is a rare monogenic migraine subtype characterised by attacks associated with transient motor weakness. Clinical information is mainly based on reports of small families with only short follow-up. Here, we document a prospective 15-year follow-up of an extended family with FHM type 2.
Patients and methods
After diagnosing FHM in a patient with severe attacks associated with coma and fever, we identified eight more family members with FHM and one with possible FHM. All family members were prospectively followed for 15 years. In total 13 clinically affected and 21 clinically non-affected family members were genetically tested and repeatedly investigated.
Results
A novel p.Arg348Pro ATP1A2 mutation was found in 14 family members: 12 with clinical FHM, one with psychomotor retardation and possible FHM, and one without FHM features. In 9/12 (75%) family members with genetically confirmed FHM, attacks were severe, long-lasting, and often associated with impaired consciousness and fever. Such attacks were frequently misdiagnosed and treated as viral meningitis or stroke. Epilepsy was reported in three family members with FHM and in the one with psychomotor retardation and possible FHM. Ataxia was not observed.
Conclusion
FHM should be considered in patients with recurrent coma and fever.
Introduction
Familial hemiplegic migraine (FHM) is a rare and clinically heterogeneous monogenic subtype of migraine with aura, characterised by attacks associated with transient motor weakness and various other neurological features (1). Three causal genes have been identified: CACNA1A (FHM1), ATP1A2 (FHM2) and SCN1A (FHM3) (2). PRRT2 has been suggested as the fourth FHM gene but its association with hemiplegic migraine is complex and needs further investigation (3).
Clinical recognition and diagnosis of hemiplegic migraine is complicated by its low prevalence and only limited phenotypic information mainly derived from cross-sectional case reports of mostly small families (4). Here we describe a 15-year clinical follow-up of an extended multigenerational FHM2 family with a novel ATP1A2 mutation and a dramatic phenotype with severe attacks of hemiplegic migraine, impaired consciousness and fever.
Methods
After clinically diagnosing hemiplegic migraine in patient IV-41 (Figure 1), all available family members were interviewed and asked to complete an extensive headache questionnaire. The family members were prospectively followed from 1997 until 2012 (15 years) with in-person or telephone interviews conducted by research physicians or trained medical students with an average time interval of five years (Table 1). Additional clinical information and diagnostic test results (e.g. from computed tomography (CT) and magnetic resonance imaging (MRI), electroencephalography (EEG) and cerebrospinal fluid analysis (CSF)) were retrieved from clinical reports of visits to out-patient clinics and admissions to hospitals throughout the Netherlands. All migraine diagnoses were made according to standard criteria (1).
Pedigree of the family with familial hemiplegic migraine (FHM) type 2. Symbols: arrow: proband; black left upper square: Migraine without aura; black right upper square: Migraine with aura; black lower half: FHM. Inner grey symbol: episodes with coma/somnolence. Question marks indicate inconclusive phenotypes. Plus ‘+’ or minus ‘–’ signs indicate the availability of a DNA sample for genetic analysis. R348P/WT indicates heterozygosity for the p.Arg348Pro ATP1A2 mutation; WT/WT indicates homozygosity for the wild-type allele. All mutation carriers suffer from hemiplegic migraine, except for IV-38 (aged 29 years at latest interview). The interview for subject IV-36 was complicated by psychomotor retardation, but she is shown as affected because she had severe attacks with hemiparalysis and epilepsy. For subject IV-44 no DNA sample was obtained. Clinical follow-up of HM attacks and associated symptoms in members of the family with familial hemiplegic migraine type 2. Aura symptoms: V: visual; S: sensory; M: motor; D: dysphasic; NA: not applicable. HM: hemiplegic migraine; TIA: transient ischaemic attack.

Because family member III-25 also developed severe restless legs syndrome (RLS) and RLS might be comorbid with migraine (5), all family members answered in 2012 a four-question telephone screener which was based on standard criteria for RLS (6). If the screener result was positive, RLS severity was assessed using the Dutch translation of a validated RLS rating scale questionnaire.
The study was approved by the Medical Ethics Committee of Leiden University Medical Centre and all participants provided written informed consent.
For genetic analyses, DNA was extracted from peripheral leucocytes in venous blood according to standard protocols. Coding regions and adjacent intronic sequences of the three FHM genes (CACNA1A, ATP1A2 and SCN1A) were initially sequenced using direct Sanger sequencing in genomic DNA of the father of the proband. After identification of the novel ATP1A2 mutation c.1043G > C, p.Arg348Pro (Figure 2(b)) in 2010, DNA of all available family members was screened for the presence of this mutation.
(a) Schematic representation of reported sporadic and familial hemiplegic migraine mutations in the ATP1A2 protein. Mutations associated with a phenotype including features of coma and/or somnolence (black circles with white numbers) are spread throughout the ATP1A2 protein, albeit with some clustering in the area around the p.Arg348Pro mutation (grey circle with number 16) at the beginning of the large intracellular loop between domain M4 and M5. Figure redrawn from De Vries et al. (2) (b) Electropherogram depicting the heterozygous c.1043G>C substitution. (c) Alignment of the amino acid sequence of homologues and orthologues of human ATP1A2, showing full conservation of amino acid Arg348.
Results
Included participants
In total 34 family members and nine spouses were interviewed and included in the analysis (pedigree in Figure 1). Family member IV-44 was interviewed but not genotyped. From family member II-6 we collected clinical information only from medical records as she already had died. Except for IV-49, all family members were interviewed on at least two occasions with average time intervals of five years (Table 1).
Genetic analysis
The ATP1A2 mutation p.Arg348Pro (c.1043G > C) co-segregated with hemiplegic migraine in 12 out of 14 (86%) mutation carriers (not including affected obligate carrier II-6) and was absent in 28 relatives without hemiplegic migraine (including nine spouses) (Figure 1). The mutation was not present in various public databases (i.e. dbSNP (http://www.ncbi.nlm.nih.gov/snp); Ensembl (http://www.ensembl.org/index.html); Leiden Open Variation Database (LOVD) (http://chromium.lovd.nl/LOVD2/variants.php?action=search_unique&select_db=ATP1A2); Exome sequencing project (ESP) (6503 samples) (http://evs.gs.washington.edu/EVS/); Genome of the Netherlands (GoNL) (769 samples) (http://www.nlgenome.nl/); 1000Genomes (http://browser.1000genomes.org)). Amino acid Arg348 is located in the large intracellular loop between transmembrane domains M4 and M5 and is fully conserved between homologues and orthologues of human ATP1A2 (Figure 2). In silico bioinformatics prediction programs all indicate mutation p.Arg348Pro as likely pathogenic: UMD predictor: pathogenic; SIFT: deleterious (score: 0); PolyPhen2: probably damaging (score: 1); Mutation Taster: disease causing (p value: 1).
Migraine attacks
Clinical characteristics of members of the family with familial hemiplegic migraine type 2 with confirmed p.Arg348Pro ATP1A2 mutation.
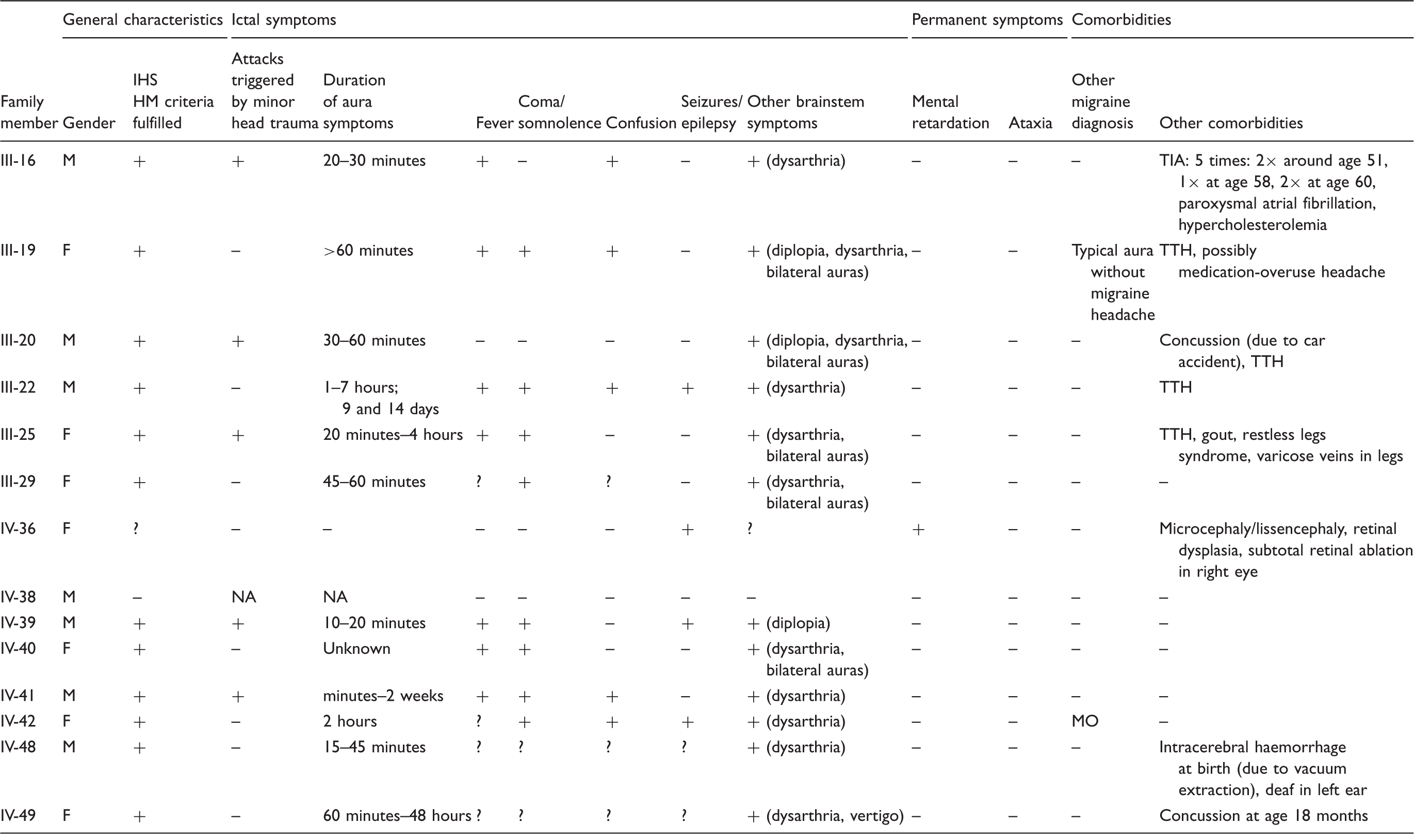
M: male; F: female; MO: migraine without aura; TTH: tension-type headache; NA: not applicable; IHS: International Headache Society; HM: hemiplegic migraine.
Subject IV-38 had never experienced migraine or other paroxysmal neurological symptoms at the time of the last interview (at age 29 years). Diagnosing whether subject IV-36 suffered from hemiplegic migraine was difficult due to severe psychomotor retardation and comorbid epilepsy. She was suspected of a lissencephaly, was wheelchair-dependent and suffered from clonic seizures with nausea and vomiting followed by hemiplegia for at least 20 minutes. These attacks appeared to be followed by headache over several days. Walking was often (increasingly) impaired for a week after such an attack.
Subject II-6 died before the study started and is an obligate ATP1A2 mutation carrier. She had several paroxysmal focal neurological symptoms during pregnancy that were (wrongly) diagnosed as transient ischaemic attacks. At age 61 she had an episode with hemiparesis and coma, with restlessness, sub-febrile temperature, and vomiting. Four years later she again developed a hemiparesis, with vertigo, yawning and dysphagia. Around age 67 to 68 she had several attacks with confusion, vomiting and hemiparesis according to her medical files.
Confusion, somnolence and coma
Nine out of 12 ATP1A2 mutation carriers with hemiplegic migraine experienced severe episodes with confusion, somnolence, or coma. These attacks often lasted days to weeks. Results from diagnostic procedures during these episodes are presented in Table 3. Abnormalities on ictal routine brain CT and/or MRI were found only in subjects IV-39 (Figure 3) and IV-41 (data not shown), in whom asymmetric congestion/hyperaemia or swelling was seen in one hemisphere. Single-photon emission computerised tomography (SPECT) showed symmetric and asymmetric areas of hypoperfusion in subjects IV-40 and IV-41 (data not shown).
Brain MRI performed during attack with somnolence and fever. Axial T2-weighted brain MRI of subject IV-39 at age 15 showing diffuse swelling of the left hemisphere (a), most evident in the left temporal region (b), with a slight rightward midline shift. Some artefacts occurred due to movements of the patient during the MRI scan. MRI: magnetic resonance imaging. Diagnostic procedures performed during or in between HM attacks in members of the family with familial hemiplegic migraine type 2. HM: hemiplegic migraine; CSF: cerebrospinal fluid; CT: computed tomography; CTA: computed tomography angiography; SPECT: single-photon emission computerised tomography; Ig: immunoglobulin; PCR: polymerase chain reaction; MRI: magnetic resonance imaging; EEG: electroencephalogram.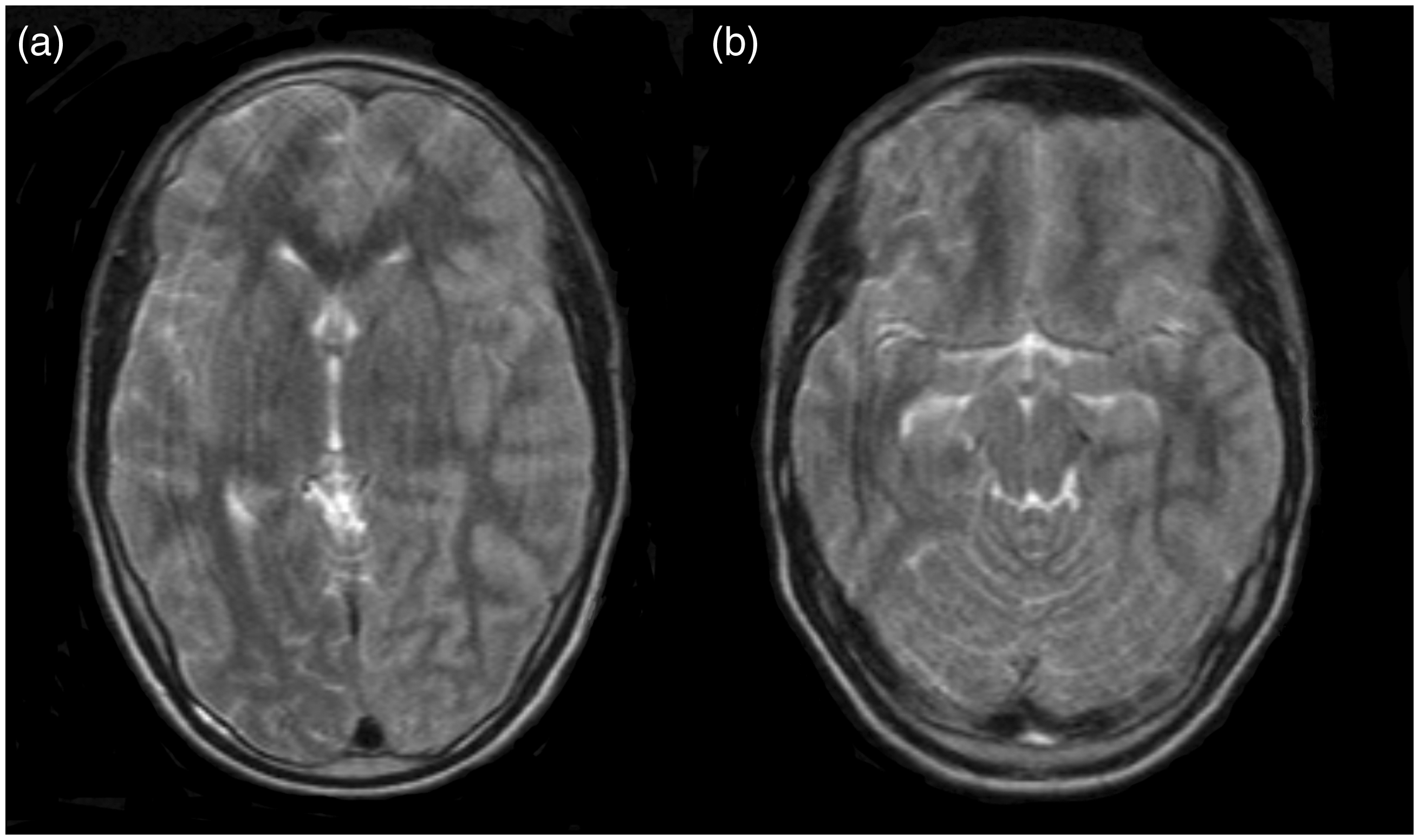

Fever up to > 39℃ was reported in seven ATP1A2 mutation carriers during severe episodes with confusion, somnolence or coma. These episodes were often suspected of viral meningo-encephalitis and treated as such. Of the nine ATP1A2 mutation carriers with severe episodes, CSF analyses were available for 10 separate episodes which revealed pleocytosis on four occasions. Viral and bacteriological tests were negative, except for one episode in subject III-19, in whom immunoglobulin (Ig)G for herpes simplex virus was reported positive in CSF and blood (polymerase chain reaction (PCR) was not yet available at the time) in combination with a high pleocytosis of 400/3 leucocytes (differentiation unavailable). She recovered spontaneously without antiviral treatment, which was not prescribed as she was pregnant at the time. CSF protein levels were within normal ranges on all occasions. A slight blood leucocytosis was observed in two patients, but C-reactive protein (CRP) levels were normal in all cases and other causes of infection were not identified during any of the severe episodes.
Epilepsy
The family members with hemiplegic migraine often described their attacks as ‘seizures’ rather than migraine but only four mutation carriers were ever diagnosed with epilepsy. EEG results are presented in Table 3. Subject IV-36 had focal epilepsy with secondary generalisation. Epileptic activity was detected on EEG at age 11, and follow-up interictal EEGs showed bilateral irritative activity, mainly in the frontotemporal regions. Subject III-22 seemingly had a seizure at age 14, when he started to ‘roll with his eyes’ and choked and gagged, but epileptiform abnormalities were not detected on repeated EEGs shortly after this attack. At age 51 he had an attack with headache, fever, confusion and agitation, and an ictal EEG initially showed only asymmetric slowing but when repeated after a few days ‘some epileptic activity’ was noted. Subject IV-39 had a single clonic seizure shortly after an attack of hemiplegia, with asymmetric severe slowing but no epileptiform abnormalities on EEG. Subject IV-42 had an apparent episode of photosensitive epilepsy around age 13 and was treated with sodium valproate for the following eight years, but epileptiform abnormalities on EEG were never reported. Non-epileptiform and (often) asymmetric EEG abnormalities with slow activity in one hemisphere were reported in eight out of 12 ATP1A2 mutation carriers with hemiplegic migraine.
Treatment
Over time, various prophylactics were used by the ATP1A2 mutation carriers with hemiplegic migraine attacks, including sodium valproate, topiramate, lamotrigine, metoprolol and phenytoin. Efficacy of prophylactic or acute treatment was not systematically reported. Medication use could not be clearly linked to reduced attack frequency, as attack frequencies were generally low. Sumatriptan was reported to have a beneficial acute effect on headache and was (retrospectively wrongly) supposed to have a beneficial effect on focal neurological symptoms during a prolonged attack in two patients (IV-40 and IV-41). Antiviral treatment (acyclovir) or antibiotics were often prescribed during attacks with coma and fever. Treatment with haloperidol did not improve symptoms of confusion (suspect of delirium) during an attack in subject III-22. Of note, subject III-25 was first suspected of cerebral infarction during a severe attack, and received intravenous thrombolysis, after which she deteriorated quickly and became comatose. She subsequently developed a high fever and was treated with antibiotics and antiviral medication. Her EEG showed marked asymmetric slowing of background activity over the left hemisphere, but MRI and CSF were normal (data not shown). She recovered slowly but fully.
Clinical course over a 15-year follow-up
The average attack frequency was consistently low, with no more than a few attacks per year (Table 1). Only two patients reported a higher attack frequency of once per month or more, but most had attack-free intervals of several years. Of note, three patients reported spontaneous exacerbations with up to three attacks a week. Several younger patients had experienced only one severe episode with somnolence, coma and/or confusion, but recurrent severe episodes were observed during follow-up in older patients (Tables 1 and 2). One subject (III-31) suffered from at least four episodes with coma. Three patients required rehabilitation therapy after attacks but eventually regained all motor skills. Several patients reported some remaining cognitive complaints after severe episodes, which had, however, not been tested formally. Overall, severe attacks were reported to occur during the entire lifespan from age 6 to 60 years. Remarkably, some patients could not accurately remember their childhood or teenage attacks which they had, however, clearly described during previous contact moments.
Clinical phenotype of relatives without ATP1A2 mutation
None of the family members without the p.Arg348Pro ATP1A2 mutation suffered from hemiplegic migraine, but some did have other migraine subtypes (Figure 1). Three (IV-33, IV-34 and IV-50) were diagnosed with migraine without aura, subject IV-33 had several attacks of probable migraine with aura and subject III-13 had a maximum of five migraine with aura attacks around age 20. Two subjects had non-motor auras without (migrainous) headache: isolated possible visual auras two to three times per year in IV-32 and visual and sensory auras two to three times per year in IV-37. DNA of subject IV-44 was not available, but she never had migraine symptoms. Like her mother (III-25), she also suffered from RLS. The ATP1A2 mutation was absent in all nine investigated spouses and none had migraine symptoms. Spouse III-17 also suffered from RLS.
Discussion
We describe a unique 15-year follow-up of a large FHM family with a novel p.Arg348Pro ATP1A2 mutation. To our knowledge, this is the largest FHM2 family with such a long prospective follow-up described to date. The long-term follow-up allowed us to observe that severe attacks with long-lasting auras and impaired consciousness, often accompanied by fever, recurred several times in the same individual, and also that none of the mutation carriers showed permanent impairments. The high prevalence of severe attacks and their recurrence in mutation carriers indicates that these are part of the FHM2 spectrum, especially since most severe attacks also included typical hemiplegic migraine aura symptoms. Importantly, some patients had only severe attacks associated with impaired consciousness and fever increasing the risk of misdiagnosis (e.g. viral meningitis). Indeed, many were not recognised as FHM patients during numerous hospital admissions. One subject was even treated with intravenous thrombolysis for suspected cerebral infarction and several patients were prescribed anti-platelet aggregating drugs because of suspected transient ischaemic attacks. Notably, several patients in this family did not consider themselves migraine patients but rather patients with transient ischaemic attacks or epilepsy. While this is not a unique feature of this family, it likely made an important contribution to the many misdiagnoses. This perception was reinforced by the relatively rare comorbidity of hemiplegic migraine with other migraine subtypes, which is in contrast to previous studies (7). However, these studies mostly included patients with clinically but not genetically confirmed hemiplegic migraine and may thus concern different genetic and clinical subtypes.
Both the strong conservation of the affected amino acid that is located in an important functional domain of ATP1A2 with many FHM2 mutations and in silico predictions strongly point towards pathogenicity of the p.Arg348Pro ATP1A2 mutation. Moreover, the mutation co-segregated with hemiplegic migraine in all 12 FHM family members. In two additional mutation carriers, hemiplegic migraine could not be unequivocally confirmed. Due to psychomotor retardation, the attacks of subject IV-36 could not be reliably differentiated from (a combination with) epilepsy (although the combination of hemiplegia, headache, nausea, and vomiting strongly suggested hemiplegic migraine). Mutation carrier IV-38 was clinically unaffected and thus could be a non-penetrant case which is not uncommon in FHM (8,9). However, because of his relatively young age (29 years old) at the time of the last interview, he still could develop hemiplegic migraine when growing older.
Characteristics of patients previously described in literature with hemiplegic migraine type 2 with episodes including somnolence or coma.

HM: hemiplegic migraine; MRI: magnetic resonance imaging; IQ: intelligence quotient; EEG: electroencephalogram.
Coma has also been described in FHM1 patients, in particular in patients with the p.Ser218Leu CACNA1A mutation (25–27). In FHM1, ictal coma has been associated with cytotoxic cerebral oedema, possibly caused by traumatic depolarisation after brain injury, when increased ionic perturbations due to the calcium channelopathy may cause cellular swelling (25,28). In our FHM2 family ictal brain MRIs are available for only a few family members, revealing unilateral diffuse swelling in two patients.
Besides a possible role for cerebral oedema, decreased consciousness in our family may have been due to spreading depression within the brainstem. In FHM1 and FHM2 transgenic mouse models, the triggering threshold for cortical spreading depression – the likely underlying mechanism for migraine aura – was decreased (29–31). In transgenic mouse models harbouring the p.Ser218Leu CACNA1A mutation, cortical spreading depression propagated into subcortical areas, basal ganglia and diencephalon and vice versa could also start in the deeper brain regions and propagate upwards (29,32). Profound subcortical spreading depression may thus explain decreased consciousness in FHM1, and conceivably also in FHM2, possibly in combination with (secondary) oedema.
Several other FHM2 families have been described with more than one patient with severe attacks associated with impaired consciousness, suggesting a true rather than coincidental association with ATP1A2 mutations (11,12,16,17,20,24). In many of these families, however, unlike in our family, comorbid generalised epileptic seizures were common (11,17,20,24). Differentiating epilepsy from hemiplegic migraine can be difficult as migraine motor auras may resemble post-ictal paresis and decreased consciousness is often seen after generalised epileptic seizures (33). However, if the paresis evolved gradually over minutes, as is typical for motor auras in migraine, these entities can be differentiated. Moreover, fever is also sometimes seen in the post-epileptic state (34). Although EEGs were frequently performed in our family, abnormalities possibly suggesting epilepsy were found in only one patient (III-28). Subject IV-36 had focal epilepsy with secondary generalisation. Slowing on EEG was reported in a number of family members which, although sometimes seen in (post-)epileptic states (35), can hardly be regarded as diagnostic for epilepsy. We would logically have expected to detect some residual epileptic abnormalities on EEG in at least a few patients if the attacks were epileptic in nature. It is important to recall that (cortical) spreading depression may show epilepsy-like electrophysiological characteristics, with recordings of spreading depressions evolving into epileptic convulsions (36). While ‘pure’ FHM without associated features may closely resemble ‘common’ migraine with aura, phenotypes on the severe end of the FHM spectrum may be more similar to epilepsy.
Paroxysmal ataxia has rarely been described in FHM2 (11,16,19,37). Although five family members reported inability to move the affected limb(s) in a coordinated manner during attacks, we believe this was due to motor weakness or loss of sensation rather than ataxia.
One spouse (III-17) and only two family members had RLS: FHM patient III-25 had severe RLS, and IV-44 had mild RLS without migraine. Therefore, although common migraine subtypes have been associated with RLS (5), we failed to find any association in our FHM2 family.
Various clinical symptoms in our family strongly resemble those of other migraine-associated syndromes. For instance, CSF pleocytosis reinforced the suspicion of (viral) infections in several patients. A headache syndrome that by definition includes CSF pleocytosis is transient headache and neurological deficits with cerebrospinal fluid lymphocytosis (HaNDL) (1). In this syndrome, as in our family, migraine(-like) headache and transient hemiparaesthesia, dysphasia, or hemiparesis can be present for several hours. Confusion, fever and uni- or bilateral slowing on EEG have also been reported in HaNDL patients but are not included in the diagnostic criteria (38–40). The only criterion excluding HaNDL in our family is that HaNDL, by definition, should resolve spontaneously within three months (1). However, follow-up in HaNDL was mostly short compared to in our FHM2 family. Only one study has screened HaNDL patients for FHM1 CACNA1A mutations, but it failed to find any in 10 patients (39). Screening of ATP1A2 or SCN1A has never been reported in HaNDL patients.
The severe episodes in our FHM2 family may also resemble acute confusional migraine, which is characterised by a reversible acute confusional state with agitation, complete or partial amnesia, speech impairment and (bi- or unilateral) slow-wave EEG changes (41). Spreading depression has been suggested as a possible mechanism, possibly in the temporal lobe or brainstem. Minor head trauma has been reported as a common trigger for confusional migraine and, as observed in our family, hemiplegic migraine. Confusional migraine and hemiplegic migraine may thus be part of the same spectrum, but with different localisation and severity. The high prevalence of brainstem auras in our family might point at a localisation in the brainstem.
In conclusion, we provide the most comprehensive report to date of the most severe end of the clinical spectrum of FHM2 characterised by recurring long-lasting episodes of hemiplegic migraine, decreased consciousness, confusion, and fever. More awareness of such severe attacks as a migraine variant is dearly needed to prevent misdiagnoses and possibly harmful treatment. The striking clinical similarities with HaNDL, confusional migraine and brainstem aura, and, from a broader perspective, similarities with epilepsy, suggest possible common underlying mechanisms.
Clinical implications
Familial hemiplegic migraine should be considered in patients with recurrent coma and fever. Clinical similarities of familial hemiplegic migraine with transient headache and neurological deficits with cerebrospinal fluid lymphocytosis (HaNDL), confusional migraine and brainstem aura, and, from a broader perspective, similarities with epilepsy, suggest possible common underlying mechanisms.
Footnotes
Acknowledgement
Author contributions are as follows: NP, acquisition and analysis of clinical/genetic data, drafting/revising the manuscript; DEB, analysis of clinical data, drafting/revising the manuscript; AHS, acquisition and analysis of clinical/genetic data, revising the manuscript; LSV, acquisition of genetic data, revising the manuscript; ATMH, acquisition of clinical data, revising the manuscript; JAvV, acquisition of clinical data, revising the manuscript; MDF, revising the manuscript, overall study supervision; AMJMvdM, revising the manuscript, genetic study supervision; JH, revising the manuscript, clinical study supervision; and GMT, revising the manuscript, clinical study supervision.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NP reports support for conference visits from Menarini. DEB, AHS, LSV, ATMH, JAvV, AMJMvdM and JH report no disclosures. MDF reports grants and consultancy or industry support from Medtronic and independent support from the European Community, the Netherlands Organisation for Scientific Research (NWO), the National Institutes of Health (NIH) and the Dutch Heart Foundation. GM Terwindt reports independent support from the NWO, the European Community, the Dutch Heart Foundation, and the Dutch Brain Foundation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NWO (VIDI 91711319 to GMT) and the European Community (FP7-EUROHEADPAIN – no. 602633 to MDF and AMJMvdM); the funding agencies had no role in the design or conduct of the study.