
Abstract
Prolonged hemiparetic migraine aura can cause diagnostic confusion and be mistaken for ischaemic stroke occurring during the course of a migraine—‘migrainous infarction’. We report a case of prolonged hemiparesis occurring during the course of a migraine attack. Though initially confused with migrainous infarction, we suggest with sequential magnetic resonance imaging, magnetic resonance angiography, diffusion, perfusion images and magnetic resonance spectroscopy that the hemiplegia was not of vascular origin and that the patient had sporadic hemiplegic migraine. We hypothesize that the mechanisms of sporadic hemiplegic migraine probably lie at a cellular level, similiar to familial hemiplegic migraine.
Case report
A 33-year-old white female presented with a confusional state and prolonged dense left-sided weakness complicating a migraine attack. She experienced migraine attacks since the age of 10. These were more prominent premenstrually. The aura symptoms comprised of teichopsia, left hemi-sensory disturbances (usually paraesthesias) and very mild weakness of left upper and lower limbs. A headache with migrainous features would accompany the aura with a total duration extending to 7 days. On this occasion she presented because of disorientation and confusion developing during day 5 of a migraine attack. Her aura was like her usual symptoms but she also reported bilateral visual field impairment and severe weakness of the left upper and lower limbs.
She was a non-smoker and did not take triptans, oral contraceptive pills or recreational drugs. There was no history of miscarriage or venous thromboembolism.
She and her mother have retinitis pigmentosa (RP). There was a strong family history of migraine with typical aura, visual and sensory, in her mother and father. No other family members had had hemiplegic attacks.
Examination showed reduced visual acuities in both eyes (6/24) with a left temporal homonymous hemianopia. She had left-sided weakness MRC (Medical Research Council) grade 3/5 in a pyramidal pattern, involving arm and leg and cortical sensory loss. She had visuo-spatial inattention for the left and anosagnosia. During her admission brief partial motor seizures of left upper limb and face were witnessed.
All blood tests including those for a prothrombotic state (antiphospholipid antibodies, protein C, S, antithrombin 3 and factor V Leiden mutations) were normal.
Magnetic resonance imaging (MRI) was done using a GE 1.5-T Signa Horizon LX Nvi MR scanner. Figures 1–6 were in the acute phase (a week into her illness) and Figs 7–12 were 3 months later. SV Proton MRS (PRESS sequence TR 1500 TE 35) studies are depicted in Figs 6 and 12.

T2-weighted FSE with slight hyperintensity and swelling of the grey matter in the right frontal, temporal, parietal and occipital regions. A small mass effect is present with mild compresssion of the third ventricle and sulcal effacement.
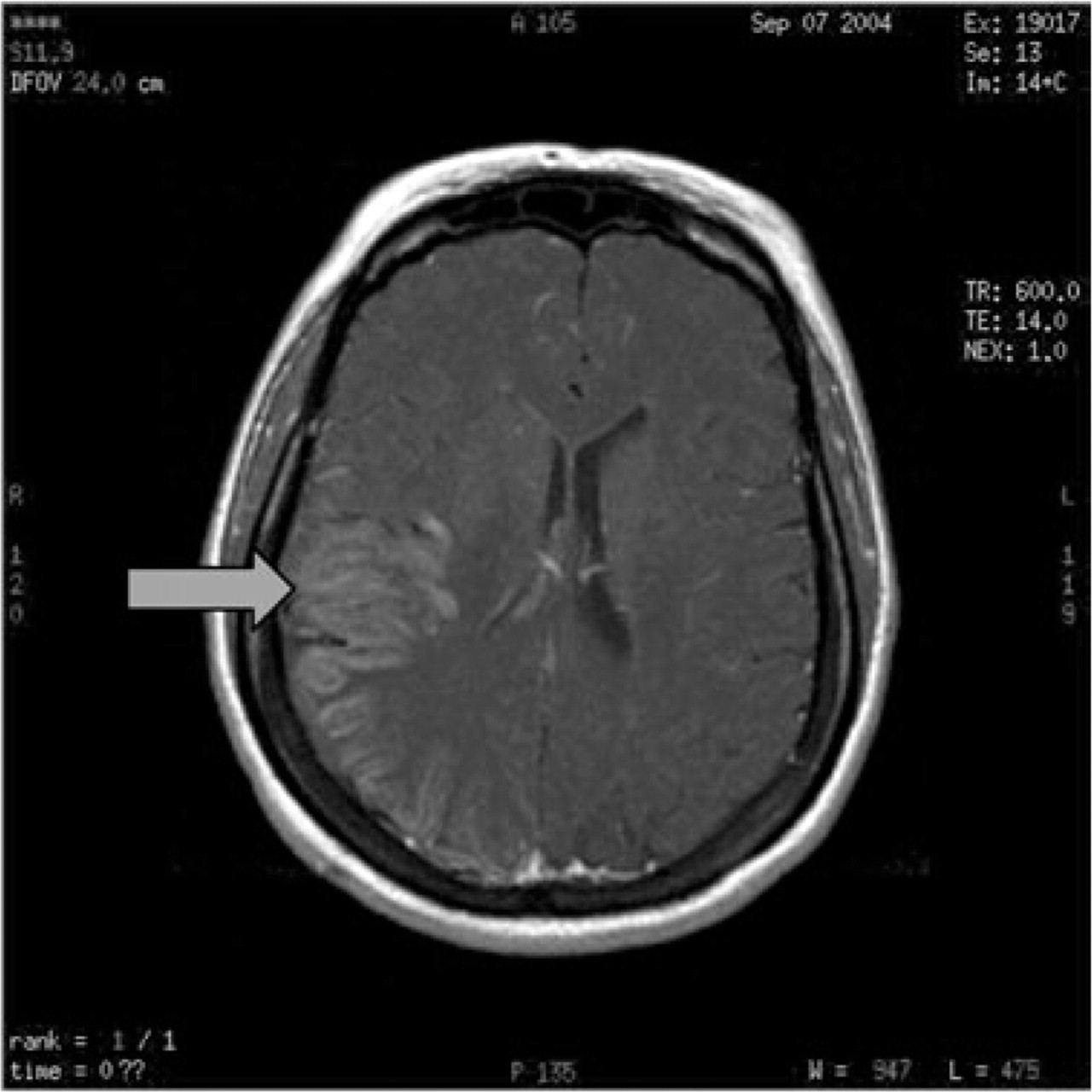
Intravenous gadolinium shows patchy enhancement of the grey matter in the affected regions.
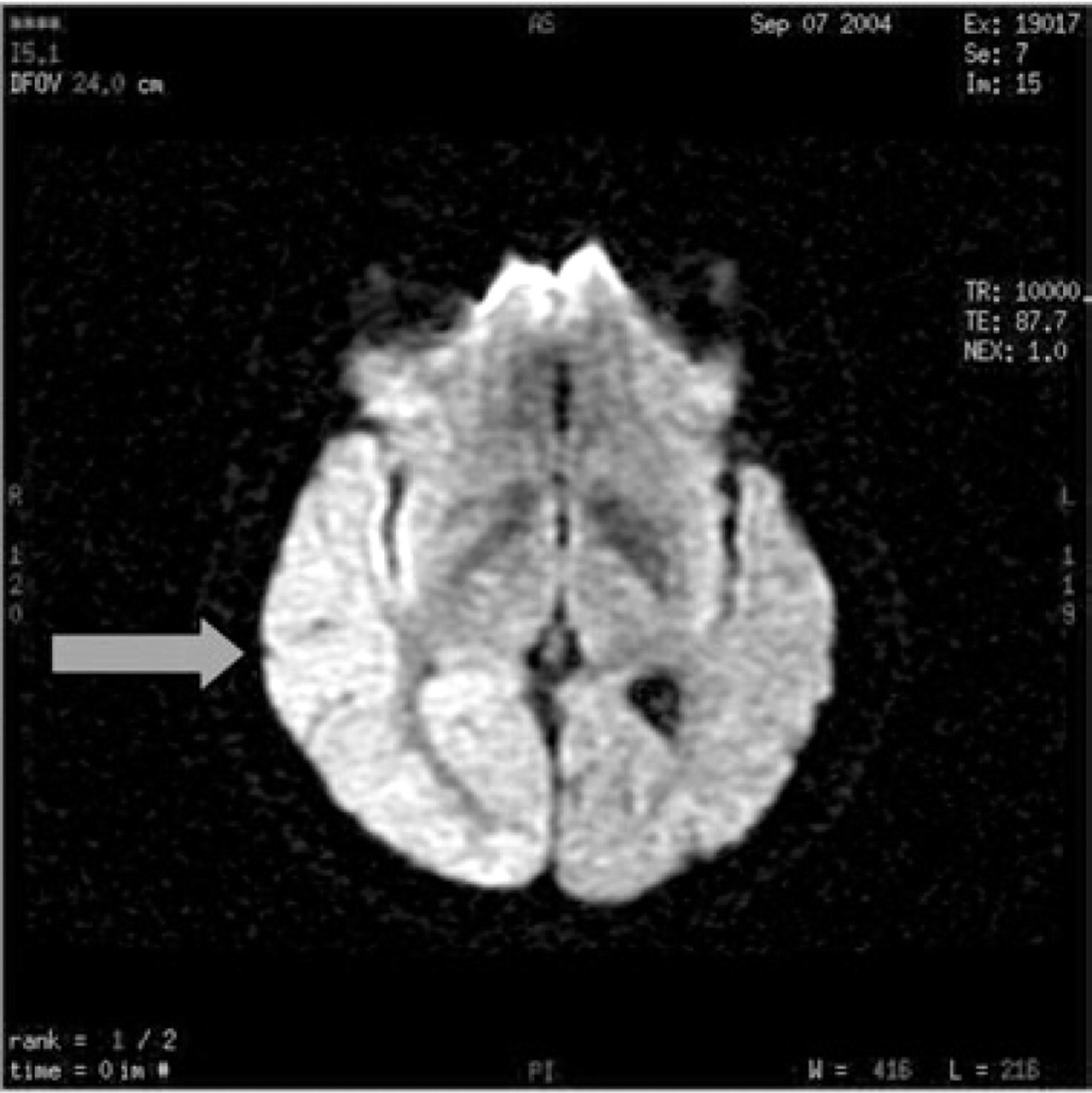
Diffusion map shows restricted diffusion in the right temporo-occipital regions.
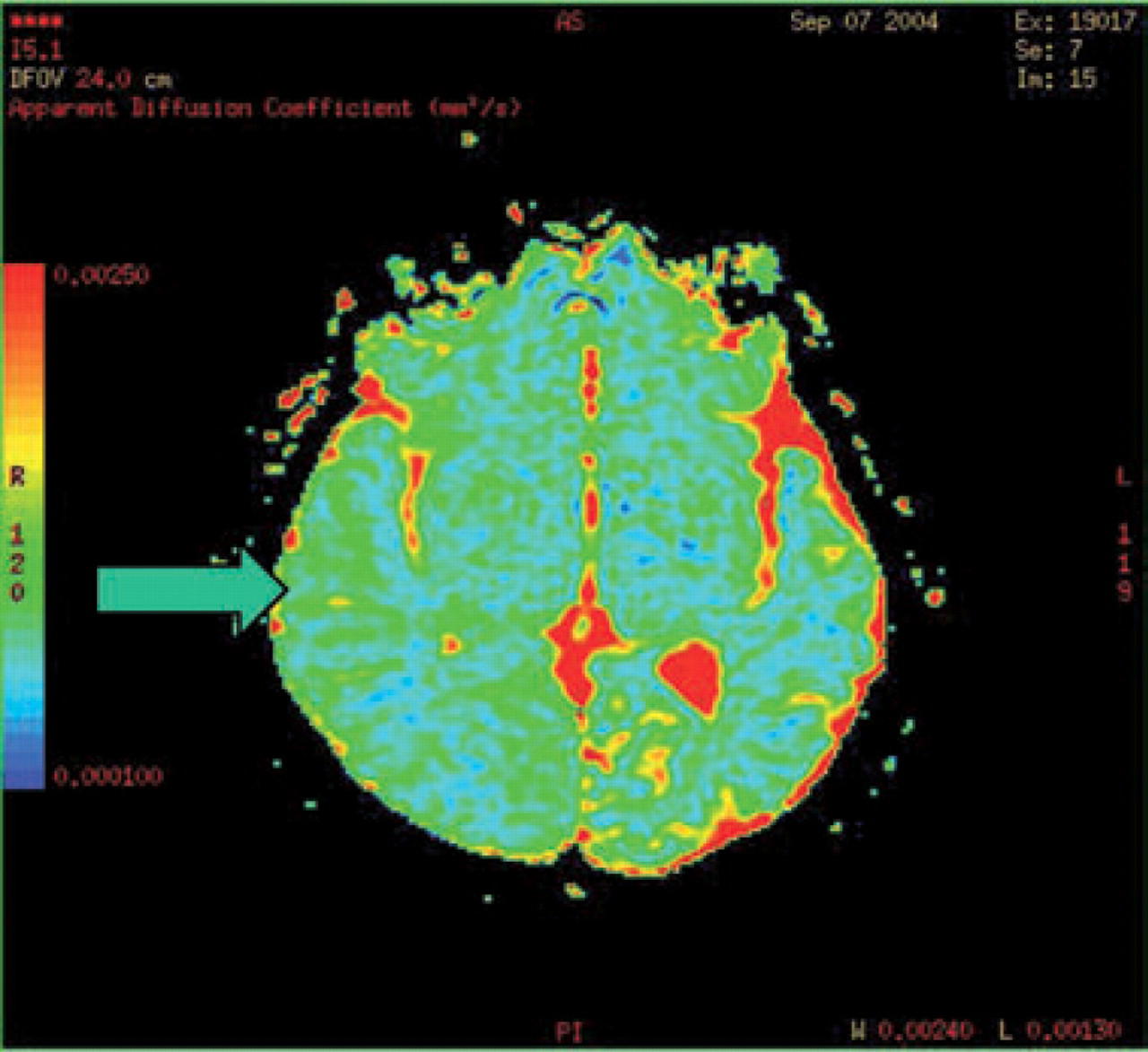
Apparent diffusion coefficient (ADC) map also shows restricted diffusion in the right temporo-occipital regions.

Dynamic susceptibility contrast MR perfusion studies show increased vascularity in the grey matter of the right temporoparietal region.

SV proton MRS (PRESS sequence TR 1500 TE 35) in a region of interest on the right side shows reduced N-acetylaspartate (NAA). Choline/creatine area ratio (Cho/Cr) is normal. Myoinositol/creatine area ratio (mI/Cr) is 0.2 (the contralateral normal side was 0.69). The findings are consistent with reduced neuronal activity. The left side (inset) appears normal.

T1 sequence shows resolution of gyral oedema after 3 months.

T2 sequence shows only minimal of gyral oedema after 3 months.

T1 with gadolinium shows no residual enhancement.

Only mild defects on diffusion mapping on the affected side, at 3 months.
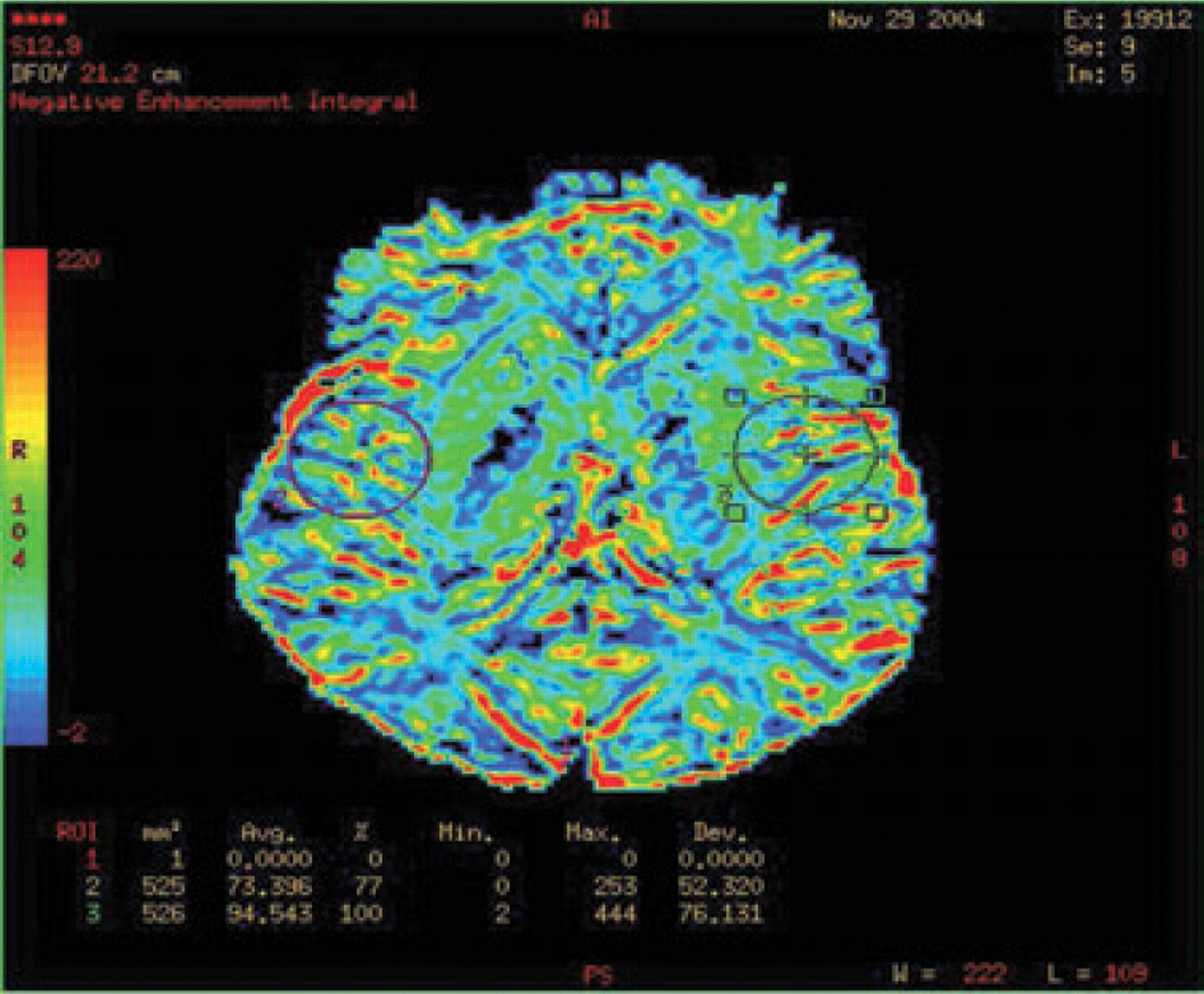
Perfusion studies at 3 months show near normal findings. There is probably slightly reduced vascularity.

The spectral pattern has reversed back to near normal levels on the affected side. The unaffected left side (inset) is shown for comparison.
MR angiography of intracranial and extracranial arteries and veins was normal. Carotid Doppler studies did not demonstrate any abnormalities to suggest dissection. EEG showed right hemispheric slow wave changes only. A transoesophageal echocardiogram was normal with no evidence of patent foramen ovale or other cardio-embolic source. Genetic tests for mutations for mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) and myoclonic epilepsy associated with ragged-red fibres (MERRF) were negative. CSF studies including lactate levels were normal. Genetic tests for the CACNA1A T666 M mutation associated with familial hemiplegic migraine (FHM) were negative.
She was managed supportively and made a complete recovery by 3 months of the initial event; repeat imaging at 3 months showed almost complete resolution. She was commenced on flunarazine 10 mg nocte and has had no further major attacks.
Discussion
We describe a 33-year-old woman who developed prolonged hemiparesis and MRI abnormalities suggestive of an infarct during the course of a migraine. These would be consistent with ‘migrainous infarction’ (1). Migrainous infarction indicates the development of a stroke during the course of a migraine and usually has involvement of large arteries. An infarct should be demonstrable by neuroimaging in a relevant region. The neurological deficit should be in the area corresponding to the origin of aura symptoms in previous migrainous episodes. Migrainous infarction can occur in those with and without risk factors such as oral contraceptive use and smoking (1, 2). However, in this patient the findings were very unlikely to be due to vascular occlusion as both the middle and posterior cerebral artery territories were involved. Also, angiography excluded any occlusion. Although there were restricted diffusion changes, perfusion studies showed hyperperfusion and these abnormalities resolved almost completely on the repeat scan at 3 months.
SV Proton magnetic resonance spectroscopy showed reduced N-acetylaspartate (NAA) on the affected side. Choline/creatine area ratio (Cho/Cr) was normal. Myoinositol/creatine area ratio (mI/Cr) was 0.2 (contralateral side 0.69). 1H 2DCSI showed reduced NAA in the affected area. These findings are consistent with a reduced neuronal metabolic activity. These abnormalities resolved almost completely on the repeat study at 3 months.
The demonstration of a fully reversible metabolic abnormality without vascular occlusion in the context of clinical migrainous-infarction indicates that the pathophysiology of this process is at the cellular level. Hemiplegic migraine is the most likely possibility. She had no family history and tested negative for the CACNA1A mutation which is the commonest in FHM (see below) We therefore believe that she has sporadic hemiplegic migraine (SHM). Very similar MRI and magnetic resonance angiographic changes have been demonstrated in FHM (3). By extrapolation, the findings in our case suggest that the mechanism underlying SHM are similar to FHM.
The most frequent inheritance of FHM is autosomal dominant. Mutations in the CACNA1A gene on chromosome 19 (FHM1) that encodes the pore forming subunit of the α1 subunit of the neuronal calcium channel account for the majority (4). A second type FHM2 is due to mutations on the ATP1A2 gene on chromosome 1 encoding for the α2 subunit of the Na/K pump (5). Recently, a third mutation on chromosome 2q24, encoding for the neuronal voltage gated sodium channel SCN1A, has been reported (6). We have not been able to test for FHM2 gene or chromosome 2q24 mutation in this patient.
SHM is described as migraine with aura including motor weakness without an affected first- or second-degree relative with aura including motor weakness (1). SHM itself seems to occur in isolation or rarely with associated cerebellar abnormalities. Three different missense mutations have been identified on the CACNA1A gene in SHM. In those with cerebellar abnormalities a mutation in the T666M and Y1384C has been identified (7). In one case of SHM without ataxia a mutation has been seen on R583Q (8).
The strong family history of migraine with aura in our patient's parents is interesting and this association has been documented previously elsewhere (9). It is also interesting that she has hereditary RP. The association of RP with FHM and migraine has been noted in a variety of disorders, including congenital disorders of glycosylation and mitochondrial disorders, but not in SHM (10–13). These disorders seem unlikely in our case.
It is conceivable from current theories of migraine that a depolarizing neuroelectric or metabolic stimulus leads to a wave of spreading depolarization causing prolonged neuronal depolarization. This could be excessive or unusually prolonged in the presence of abnormal calcium or sodium channels. The altered membrane permeability and ensuing dysfunction might be clinically evident as auras or hemiplegia (2, 14). The reduced diffusion is probably related to cell swelling due to failure of ATPase pump (15). These mechanisms that are proposed to operate in FHM subtypes may occur in SHM also. Anecdotal effectiveness of verapamil and flunarazine, both calcium channel blockers, may suggest similarities between the pathophysiology of the conditions (16–18).