
Abstract
Background: The clinical manifestations of sickle cell disease (SCD) vary, but may be attributed to vaso-occlusion, chronic hemolytic anemia, and infections as a result of functional asplenia. We report a case of a man who presented with severe headache caused by an uncommon complication of SCD.
Case: A 19-year-old Surinamer man presented to the emergency department with severe headache. The progressive headache started one day previously. The headache was located frontotemporally on the right side. It was pulsating with paroxysms of fierce pain. There was no nausea or vomiting. The medical history reported sickle cell disease of the HbSC type. The physical and neurological examination was normal. He was afebrile with a blood pressure of 118/72 mmHg. Blood tests and CSF investigation showed no abnormalities. CT-scan of the head was normal. The headache disappeared after two days. Eight days later he presented again, with a relapsing severe headache. Physical, neurological examination and blood investigations were normal. MRI now showed infarction located in the parietal skull bone, with a small adjacent epidural hematoma. The headache disappeared gradually over 8 days. Repeat MRI one month later showed complete disappearance of the epidural hematoma. The first headache episode was thought to be due to the initial skull bone infarction as no epidural hematoma had been present initially. The second headache episode was thought to be due to the development of the epidural hematoma.
Discussion: A skull bone infarction is an uncommon complication of SCD, as typically these are located in the long bones. Even more uncommon is a epidural hematoma which was probably the result of the altered bone and vesselwall structure following the skull bone infarction. To our knowledge this is the first case reporting a skull-bone infarction with adjacent spontaneous epidural hematoma in an adult with sickle cell disease of the HbSC type. Our case emphasizes the need to recognize skull infarction and a concomitant spontaneous epidural hematoma as a possible complication of SCD.
Introduction
Sickle cell disease (SCD) is an inherited haemoglobinopathy characterized by the presence of sickle-shaped red blood cells that can affect any organ system. The clinical manifestations of SCD vary, but may be attributed to vaso-occlusion, chronic haemolytic anaemia and infections as a result of functional asplenia. We present a patient with severe headache due to an uncommon manifestation of SCD.
Case report
A 19-year-old Surinamese man was referred to our emergency ward with severe headache. The headache had been present when he woke up and lasted several hours. He used acetaminophen and a NSAID. At presentation, the headache was located on the right parieto-temporal side of the head and was continuous with a fluctuating intensity, with bouts of severe pain lasting for 15 minutes followed by short intervals of several minutes with less pain. It had a pulsating character with intermittent stabbing pain. There were no other accompanying signs, besides slight conjunctival injection of the eyes.
He had a history of sickle cell disease of the sickle-haemoglobin C disease (HbSC) type, with few vaso-occlusive crises, and used folic acid to treat the anaemia. He reported no previous history of headaches.
The results of physical and neurological examinations were normal, including normal blood pressure and temperature, and no signs of meningism or papilloedema. The results of a full blood count and a cerebrospinal fluid examination were normal. A computed tomography (CT) scan of the head did not show any abnormalities except for the thickened skull that is often found in SCD owing to increased haematopoiesis (Figure 1).
The initial CT scan, in which no explanation of the headache was found.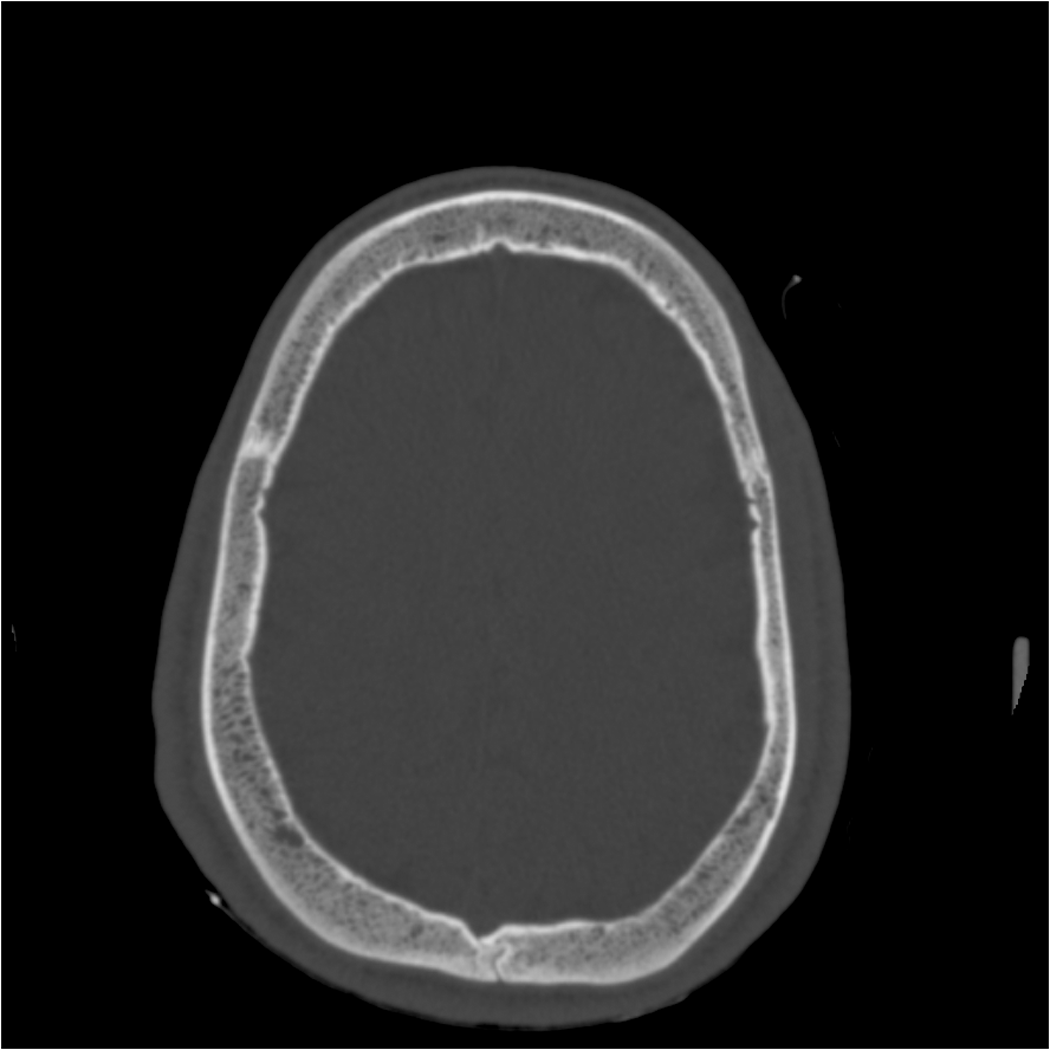
He was treated with 100% oxygen at 7 l/min for possible (atypical) cluster headache, which did not help. After this initial treatment, oral indomethacin 75 mg was given, as paroxysmal hemicrania was suggested, also without marked effect. As the headache persisted and could not be classified as a primary headache disorder the patient was admitted to the neurology department. The next day the headache had disappeared and he was discharged from the hospital.
Four days later he presented again to the emergency ward with a non-acute, progressive headache, which started the day after dismissal. He now reported having his worst headache ever, which was of a stinging character and located in the temporal-frontal region bilaterally. Photophobia, phonophobia, nausea, vomiting and autonomous signs were all absent. Physical and neurological examination were again normal. There were no local signs of infection or inflammation. Laboratory investigations did not show any abnormalities.
A magnetic resonance imaging (MRI) scan made during his second admission revealed a small, 9 mm epidural haematoma located right parietal with a hyper-intense signal of the overlying skull suggesting bone infarction or osteomyelitis (Figure 2). The epidural haematoma and the bone infarction were treated conservatively, and the headache was treated only with common analgesics. The headache fluctuated in intensity and resolved after 7 days. MRI was repeated after 1 month, and showed complete resorption of the small epidural haematoma; the overlying skull bone still showed signs of the parietal bone infarction (Figure 3).
MRI scan showing a right parietal hyper-intense signal of the skull, suggesting bone infarction, and an adjacent small epidural haematoma. After a month, the epidural haematoma is no longer present. The right parietal bone infarction is more evident.

Discussion
We report a case of a 19-year-old man who presented with two episodes of headache shortly after each other, who was found to have a skull bone infarction with a small adjacent epidural haematoma. We suggest that the first episode of right parietal headache was due to the underlying skull bone infarction. The intact bone cortex on imaging as well as the absence of fever and signs of infection in the blood made osteomyelitis less likely. The second headache episode two days later was probably due to the development of the non-traumatic epidural haematoma as a result of altered bone and periosteal structures, and this headache persisted for 7days.
The first headache episode did not fit any primary headache disorder, but a first episode of trigeminal autonomic cephalalgia was considered, because of the short severe bouts lasting 15 minutes in combination with the conjunctival injection of both eyes. However, as no other or clear unilateral autonomic symptoms were present, the headache is best regarded as a headache not elsewhere classified.
The second episode with bilateral headache without autonomic or vegetative symptoms was initially best classified as a headache not elsewhere classified. Although the bilateral localization without other symptoms suggested tension type headache the severity and stinging character were atypical. After repeat imaging the epidural haematoma was found, and the headache was attributed to the epidural haematoma. Focal neurological signs were absent however. For headache attributed to epidural haematoma no typical characteristics are known according to the ICHD-II classification.
Headache is relatively common in children and young adults with SCD (1). Causes of headaches in SCD are probably heterogeneous, both primary, such as migraine and tension-type headache, and secondary to severe anaemia. Recently, increased transcranial Doppler velocities were found in migraine-mimicking headache in SCD (2). Bone infarctions are a common clinical manifestation of SCD, which are mainly located in the long bones, ribs and spine (3). The skull bone is a less common location for infarction. Bone marrow is highly vascularized and therefore vulnerable for infarction. Bone infarctions in the skull bone are most frequently reported to be located in the orbital wall, the mandible and the skull base (4).
Differentiating bone infarction from osteomyelitis radiologically can be problematic (5), as imaging findings of osteomyelitis are similar to bone infarction and include bone marrow oedema, subperiosteal fluid collection and abnormal enhancement of adjacent soft tissues (6). Cortical defects may indicate osteomyelitis (7).
Case reports describing both epidural haematomas and skull bone infarctions in sickle cell disease (SCD) patients

The exact mechanism for the epidural haematoma is unknown, but probably it is related to the skull infarction. The most likely pathophysiological mechanism is bleeding into the epidural space owing to the disrupted bone margins and vessel walls caused by vaso-occlusion. (8). All cases reported previously were in homozygous (HbSS) or unknown type of SCD. To our knowledge this is the first case reporting a skull-bone infarction with epidural haematoma in a patient with SCD of the HbSC type (Table 1). Our case and others emphasize the need to recognize skull infarction, possibly resulting in epidural haematomas, as possible causes of headache in SCD patients.