
Abstract
Background: It has been proposed that TRPV1 receptors may play a role modulating trigeminal sensory processing. We used models of trigeminovascular nociceptive activation to study the involvement of TRPV1 receptors in the rat. Due to a possible role of TRPV1 receptors in cortical spreading depression (CSD), an experimental phenomenon sharing many features with migraine aura, we also utilized a model of mechanically induced CSD.
Methods: Male Sprague Dawley rats (N = 39) were anesthetized and cannulated for monitoring and drug administration to study the effects of the TRPV1 receptor antagonist A-993610 (8 mg kg−1 IV). Wide-dynamic-range neurons, responding to electrical stimulation of the middle meningeal artery (MMA)/dura mater were identified and recorded using electrophysiological techniques. Intravital microscopy was used to study neurogenic dural vasodilation (NDV) of the MMA comparing capsaicin and electrical stimulation, and the effect of A-993610 on mechanically induced CSD was examined.
Results: Administration of A-993610 had no significant effect on trigeminal firing of A- or C-fibers elicited by electrical stimulation of the MMA. It also showed no effect on NDV whilst blocking vasodilation due to intravenous capsaicin injection. The mechanically induced CSD response could not be altered by A-993610 administration.
Conclusions: Although there is evidence that TRPV1 receptors play an important role in sensory processing in general, the new data do not support a role in the treatment of acute migraine.
Introduction
Migraine is a highly disabling (1), common (2) and expensive (3,4) brain disorder (5). The development of triptans, serotonin 5-HT1B/1D receptor agonists (6), was a substantial advance in acute migraine therapy, although only one-third of patients are headache-free two hours after treatment (7). Moreover, there are important contraindications to their use in the setting of cardiovascular and cerebrovascular disease (8). New medicines in development have been directed at neural targets (9), and their efficacy, such as is demonstrated for calcitonin gene-related peptide (CGRP) receptor antagonists (10), demonstrate that such a mechanistic approach is plausible. One suggested target for possible drug interaction in the pathophysiology of migraine is the transient receptor potential cation channel, subfamily V, member 1 (TRPV1) receptor.
TRPV1 receptors functioning as non-selective cation channel can be activated by capsaicin, protons, noxious heat (>43°C), products of cyclo-oxygenase and lipo-oxygenase, as well as different venoms from spiders and many more compounds (11–15). It has been shown to be involved in peripheral pain perception, and the many avenues of receptor activation, as well as the finding of central TRPV1 receptors, suggest a possible potential therapeutic target. TRPV1 channel activation leads to release of CGRP, which is capable of triggering trigeminal firing (16).
Trigeminovascular activation is believed to be involved in the pathophysiology of migraine. Electrical stimulation of meningeal structures is painful in humans (17) and animal models, utilizing the electrical stimulation of these structures have shown to be a valuable tool in predicting possible drug effects on the trigeminal activation (18). Using electrophysiological techniques this activation can be recorded from second order neurons in the trigeminocervical complex (19). Another model that is widely accepted due to its high predictability in headache therapeutics is intravital microscopy (20). This relies on the observation that stimulation of dural afferents produces vasodilation that can be monitored in animals.
About 30% of migraine attacks feature an aura characterized as a focal neurological deficit developing within five to 20 minutes and typically lasting maximally 60 minutes (21). The aura has been shown to have its physiological equivalent in a wave of depolarization, which is spreading across the cortex at 2–6 mm min−1 as cortical spreading depression (CSD) (22). Due to neurovascular coupling cerebral blood flow changes seen with CSD can be directly measured.
Here we utilized the models of trigeminal activation and electrophysiological measurements in the trigeminocervical complex, and the model of CSD induced by needle plunge (NP) to explore TRPV1 mechanisms potentially relevant to migraine. The data have been reported in preliminary form (23,24).
Methods
Common preparation
All experiments were conducted under license of the University of California, San Francisco (UCSF), Institutional Animal Care And Use Committee, and conforming the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments adhered to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain (25).
Male Sprague Dawley rats (270–380 g) were anesthetized with sodium pentobarbital IP (60 mg kg−1). The left femoral artery and vein were cannulated for blood pressure (BP) monitoring (CT-1000 + PM-1000 + ALM 932, CWE, Inc., USA) and further administration of anesthetics (propofol 20–25 mg kg−1h−1 and for all intravital microscopy experiments pentobarbital 18 mg kg−1h−1 was used); the right femoral vein was cannulated for drug administration. Pentobarbital was used as an anesthetic for the intravital experiments to enable comparisons with previous laboratory data (26). Animals were cannulated with a tracheal tube for ventilation with oxygen enriched air, 2–3 ml, 80–100 strokes min−1 (Small rodent ventilator–model 683, Harvard Instruments, UK). End-tidal CO2 was monitored (Capstar – 100, CWE, Inc., USA) and kept between 3.5–4.5%. Anesthesia was then maintained by propofol (20–25 mg kg−1 h−1) or pentobarbital (18 mg kg−1 h−1). Temperature was kept in physiological range via a thermostatically controlled homeothermic blanket system. Animals were fixed in a stereotactic frame for the following surgery and recordings. BP, end-tidal CO2 and temperature were constantly displayed. The withdrawal reflex after paw pinch and testing of the corneal reflexes was used to monitor the depth of anesthesia. At the end of the experiment animals were euthanized by a lethal dose of pentobarbital. Data for all experiments were displayed and saved on a personal computer using an online data analysis system (Power 1401plus, CED and Spike5 software, UK).
Trigeminocervical-complex recording
A midline incision was made reaching from bregma to C3. The parietal bone above the middle meningeal artery (MMA) was then removed using a saline cooled dental drill. A platinum stimulating electrode (NE 200X, Clark Electromedical, Reading, UK) was then placed on the dura mater next to the MMA for electrical stimulation. For recordings from the trigeminocervical complex a C1 hemilaminectomy was performed and the dura mater was removed above the site of electrode plunge. The recording electrode (0.5 MΩ tungsten recording electrode, UK, tip diameter 0.5 µm, World Precision Instruments) was lowered into the trigeminocervical complex using a piezoelectric motor/controller system (IW-811, Burleigh Instruments, Harpenden, UK; 8200 Controller, EXFO, Plano, TX, USA) in 5-µm steps. Wide-dynamic-range neurons, identified by noxious pinch, and innocuous brush, responding to electrical stimulation of the middle meningeal artery/dura mater (MMA; stimulation parameters: 0.5 Hz, 0.3–0.4 ms, 12.6–18 V), were identified and recorded (Figure 1B). Post-stimulus histograms (PSTHs) were established for twenty stimulations (Figure 1C). A-993610 (8 mg kg−1) (27), dissolved in polyethylene glycol (PEG) 300 (8 mg ml−1), or the vehicle (PEG 300) 1 ml kg−1, were administered intravenously. Prior to administration of the drug/vehicle baseline PSTHs (mean out of four stimulation series of 20 sweeps) were evaluated, and these were further collected 5, 10, 15, 20, 25, 30, 45, 60, 75 and 90 minutes postadministration. Receptive fields within the V1/V2 branch of the trigeminal nerve were tested repetitively for a duration of 3 seconds by gentle brush with a blunt probe, representing a non-noxious stimulus, and toothed forceps, representing a noxious stimulus. The testing of the receptive fields was repeated at 30, 45, 60, 75 and 90 minutes postadministration.
Summary of data from electrophysiological measurements in the trigeminocervical complex. (A) Left side: location of lesion sites in the trigeminocervical complex. Right side: example of a histological picture for a lesion site (as indicated by circle) in the dorsal horn. Staining was obtained by neutral red. (B) Trace of second order neuron cluster response to electrical stimulation of the middle meningeal artery (MMA) with the superimposition of three original traces. Responses of A- and C-fibers can be seen. (C) The associated post-stimulus histogram of neuronal response to electrical stimulation of the MMA (ordinate displays the number of units firing over 20 sweeps. (D, E) Display of responses to electrical stimulation of the dura/MMA in relation to time after administration of A-993610 (8 mg kg−1) or vehicle separated for A-fiber mediated responses (D) and C-fiber responses (E) (values for 0 indicate mean baseline responses). (F) Display of cell firing responses due to noxious and non-noxious stimulation of the receptive field in relation to time after administration of A-993610 (8 mg kg−1) or vehicle (values for 0 indicate mean baseline responses). s = seconds. ms = milliseconds. min = minutes.
According to anatomical measurements and nerve conduction velocities all recorded responses meeting criteria for classification as A-fibers (response 4–20 ms post-stimulation) or C-fibers (20–100 ms post-stimulation).
At the end of the experiment the recording site was electrically lesioned and tissue was collected for further histological processing. Topological localization of lesion sites was then identified according to the atlas of Paxinos and Watson (28).
Intravital microscopy
After exposing the skull, the parietal bone of the right or left side was thinned by using a saline-cooled dental drill until the underlying vessels of the dura mater were clearly visible and the skull was left intact (closed). The area was then covered with mineral oil (37°C) and an intravital microscope (Microvision MV2100, UK) was then focused on a branch of the MMA and projected on a TV monitor. Online monitoring of the blood vessel diameter was achieved through a video dimension analyzer (Living System Instrumentation, USA). After a resting period and evaluation of a steady vessel diameter, one of the following protocols applied.
Capsaicin-induced dilation. Capsaicin (15 µg kg−1 dissolved in tween 80 : ethanol : saline; 1 : 1: 8) was administered intravenously. The dose was previously tested to induce the maximal dilatory effect. Injection of capsaicin was repeated 15 minutes after the first injection. To allow tracking of changes in the vessel diameter due to drug or vehicle administration, either 8 mg kg−1 of the drug (A-993610 dissolved in polyethylene glycol 300) or the corresponding volume of the vehicle (1 ml kg−1) was injected IV after a recovery period of 15 minutes. The injection of capsaicin was then repeated 15 and 30 minutes postdrug administration.
Neurogenic dural vasodilation. A bipolar stimulation electrode (NE 200X, Clark Electromedical) was placed within a range of about 200 µm to the monitored vessel. The settings for stimulating the bone adjacent to the middle meningeal artery were adjusted to 5 Hz, 1 ms for 10 s (Grass Stimulator S88, Grass Instrumentation, USA). For each animal the voltage was increased until a maximal dilation was observed, all following stimulations were then performed using this voltage (29,30).
Cortical spreading depression
Anterior to bregma the frontal bone was removed in an area of approximately 2 × 2 mm using a saline cooled dental drill. This area later was the site where CSD was induced by mechanically lesioning the cortex by a needle plunge of a 26-gauge needle that was fixed in a micromanipulator. Frontal to lambda an area the same size was thinned using a saline-cooled dental drill until the dural vessels were clearly visible and the parietal bone was still intact. Posterior to this closed bone-window and in line to the frontal burr hole, a small hole was drilled into the skull and the dura mater was removed. Using a hydraulic microdrive, a NaCl (3M) filled single barrel glass microelectrode with a tip diameter of 2 µm was then inserted 800–1100 µm into the cortex for cortical steady state potential recording (direct current [DC]shift). The fluid filled electrode was connected to an Ag/AgCl pellet and an Ag/AgCl reference electrode that was placed in the neck. The electrode was connected to electrical amplifiers and to a 60-Hz noise eliminator which fed into a gated window discriminator. The signal was passed through an analog to digital converter (Cambridge Electronic Design, UK) to be displayed and saved on a PC (using Spike5v2 software). Two control CSDs 45 minutes apart were induced by mechanical stimulation of the cortex. Twenty-five minutes after the second CSD the drug vehicle was given in an equal volume as drug (polyethyleneglycol 300 1 ml kg−1), and 20 minutes later another needle stick was performed. The resulting third CSD was followed by IV drug administration of A-993610 8 mg kg−1 after which mechanical stimulations were performed at 20 and 65 minutes postadministration of A-993610.
All measurements for burr hole positions, and positioning of instrumentation in and above the cortex, were recorded for later analysis.
Statistics
All statistical analysis was carried out using SPSS (16.0, IL, USA). Statistical analysis was performed using Analysis of Variance (ANOVA) for repeated measures. If assumption of sphericity was violated Greenhouse-Geisser corrections were applied. Bonferroni correction was applied for multiple comparisons if applicable. Statistical significance was set at p < .05. In case of p values < .05 post-hoc comparisons were made using paired-sample t-test for the effect of each intervention. Additional independent t-tests were used for group comparisons of the different interventions. Results are expressed as percentages of baseline.
Results
Trigeminocervical-complex recording
In total recordings were made from 11 cells within laminae III–V of the trigeminocervical complex (Figure 1A). Ten cells displayed firing latencies consistent with A-fiber input and five of these cells also showed firing latencies consistent with C-fiber input (Figures 1B and 1C). Only one of the cells received inputs restricted to C-fiber stimulation.
Dural afferents: Administration of A-993610 had no significant effect on trigeminal A-fiber firing (F 2.6,21.2 = 0.90; p = .45; Figure 1D) and no significant effect on C-fiber firing (F 3.3,19.7 = 2.38; p = .09; Figure 1E) due to electrical stimulation of the MMA. The equivalent volume of vehicle had no significant effect on A- or C-fiber firing.
Cutaneous receptive fields: Non-noxious stimulus triggered a mean firing of 200 ± 19 Hz and noxious stimulation a firing rate of 292 ± 25 Hz. Neither the drug nor the vehicle application significantly changed the discharge rate of either stimulus (p ≥ 0.34; Figure 1F).
Blood pressure: Injection of the drug, as well as of vehicle control, led to a BP response with a significant decrease of 20% compared to baseline BP (drug: t
9 = 6.05, p < .001; vehicle: t
9 = 4.58, p = .001), followed by a significant increase of 29% compared to baseline BP (drug: t
9 = 6.52, p < .001; vehicle: t
9 = 5.35, p < .001). There was no significant difference between drug and vehicle (Figure 2).
Summary of blood pressure (BP) response to intravenous application of A-993610 (8 mg kg−1) in polyethyleneglycol 300 (8 mg ml−1) or the according volume of the vehicle. min = minimum. max = maximum. inj = injection. s = seconds.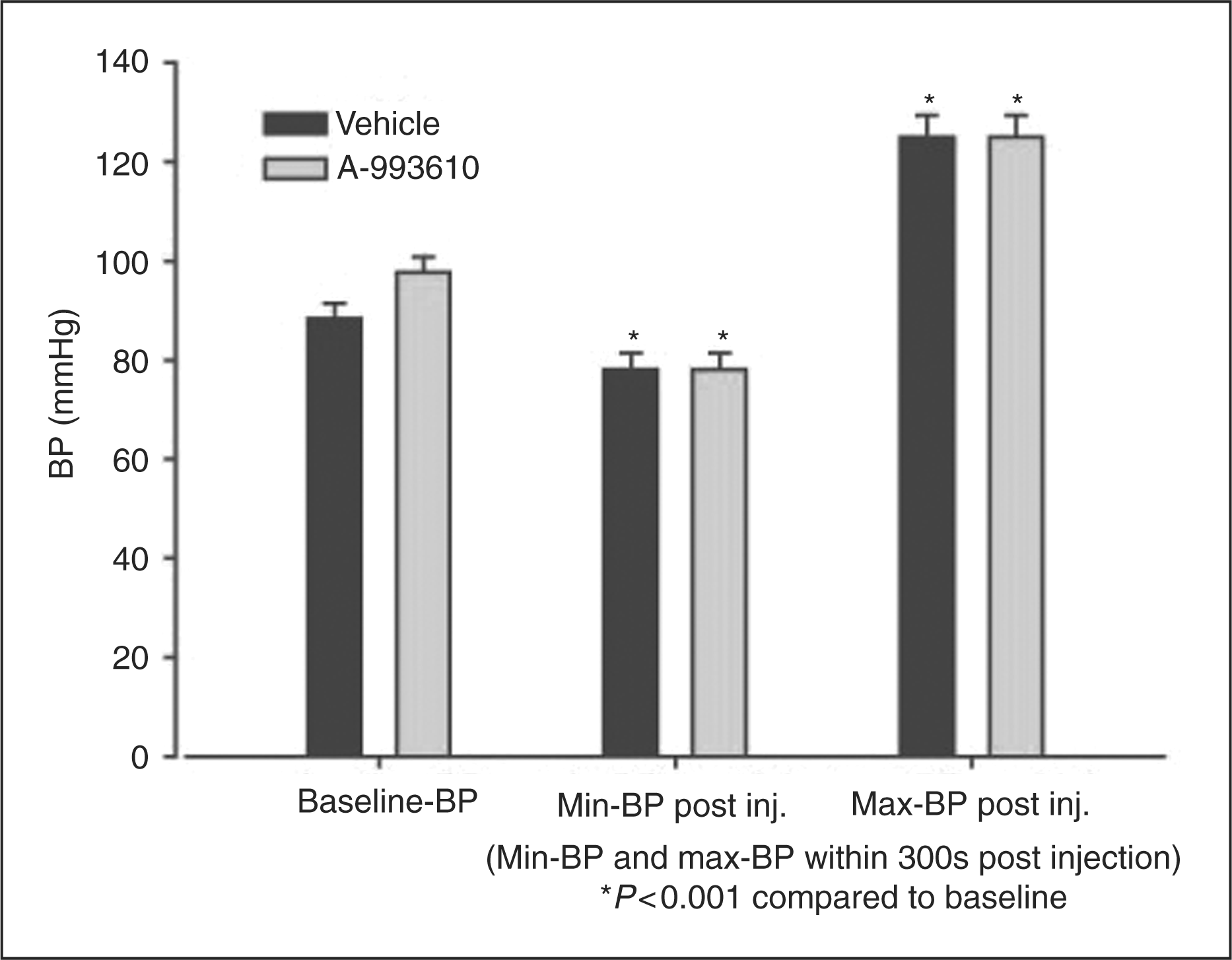
Intravital microscopy
Capsaicin-induced dilation
The effect of A-993610 on capsaicin-induced vasodilation was studied in 10 animals.
Capsaicin and blood pressure
Capsaicin administration resulted in a triphasic BP response in which an initial drop of 21% of BP at rest is followed by a short-lasting elevation of 17% of BP at rest, and then a prolonged decrease of the blood pressure of 21% BP at rest (Figure 3A). The initial BP value was reached again after about 180 seconds. This sequence of BP changes was reflected in the measurements of the vessel diameter in which an initial short-lasting decrease below the methods detection limit was followed by a prolonged 81% dilation at rest (t
9 = 13.36; p < .001).
Summary of responses observed using intravital microscopy. (A) Baseline blood pressure (BP) and vascular diameter (VD) response to intravenous application of capsaicin (15 µg kg−1) in Tween 80: ethanol: saline (1 : 1 : 8). (B) Capsaicin-induced response 15 minutes after vehicle (intravenous polyethyleneglycol 300 (1 ml kg−1) administration. (C) Capsaicin-induced response 15 minutes after administration of A-993610 (8 mg kg−1) in polyethyleneglycol 300 (8 mg ml−1). (D) Summary of VD responses to neurogenic vasodilation (time is displayed relative to injection of the A-993610 or vehicle at time point 0). min = minutes. s = seconds.
The BP response to intravenous administration of either A-993610 or vehicle is reported above and a congruent, significant but mild, vasoconstriction of 20% with full recovery of baseline before capsaicin was injected again was observed.
BP and dural vascular diameter to capsaicin injection were not altered by administration of vehicle (Figure 3B). After administration of A-993610 the BP reaction to capsaicin injection showed a short-lasting decrease, significantly different from baseline condition (F 1.05,4,21 = 14.29, p = .016) followed by a prolonged increase not significant different from baseline condition (Figure 3C). The third component of BP reaction to capsaicin injection as seen in the baseline condition (a decrease) could not be observed at all. The initial decrease of the BP induced by capsaicin was 50% lower, 15 minutes after administration (t 4 = 3.80; p = .019) and 30 minutes after administration (t 4 = 3.84; p = .019) of A-993610, than the effect measured pre–drug administration.
Capsaicin-induced dural vasodilation: There was a significant difference in the dural vascular response to capsaicin injection after A-993610 administration (F 1.1,4.3 = 102.6, p < .001). Instead of the initial substantial decrease seen in control studies, a vasoconstriction of 21% at rest (t4 = 10.98; p < .001) that lasted for about 290 seconds was observed. The dilation of the vessel observed in the baseline and vehicle condition was blocked. The effect did not differ 15 or 30 minutes post–A-993610 administration.
Neurogenic dural vasodilation
Twelve rats were utilized to study any effect of A-993610 on neurogenic dural vasodilation. The electric current needed to induce maximal dilation ranged from 100 to 170 µA. Vessel diameter changes after electrical stimulation were significant for all time points (p ≤ .002). Electrical stimulation induced an 84% dilation of baseline diameter (t 11 = 14.72; p < .001). The administration of either drug or vehicle had no significant effect on this dilation (p ≥ .24) (Figure 3D). As seen in the capsaicin-induced vasodilation experiments, the administration of A-993610 or vehicle induced a significant but completely reversible vasoconstriction.
Cortical spreading depression
Needle stick to the cortex consistently induced an increase in blood flow and a negative DC shift consistent with CSD (Figure 4). Every DC shift recorded was preceded by a blood flow increase. After administration of A-993610 needle stick still induced a CSD consistently. Drug vehicle alone also showed no effect on occurrence of CSD. There was no significant change of the onset of the blood flow change (p = .2) whilst there was a change in the onset of DC-shift (F
3,15 = 5.73, p < .05). However, the following t-test revealed the observed effect was not specific to drug administration (t
5 = −3.624, p < .05) but also seen after the administration of vehicle (t
5 = −3.318, p < .05). The effect observed after either drug or vehicle administration were not different from each other (p = .35). Throughout the recordings, with the experimental setup used, the changes in blood flow preceded the DC shift, and therefore no change in directionality can be assumed.
Typical changes seen during mechanically induced cortical spreading depression. NP = needle plunge. LDF = laser Doppler flow. BP = blood pressure. Amp = amplitude. s = seconds. drug = intravenous A-993610 8 mg kg−1 in polyethyleneglycol 300 [8 mg ml−1]. vehicle = intravenous polyethyleneglycol 300 1 ml kg−1.
Discussion
The data from the present study do not support a significant role for the TRPV1 receptor in either nociceptive trigeminovascular signaling through the trigeminocervical complex or in neurogenic dural vasodilation. The data are consistent with a previous study utilizing intravital microscopy and a less-potent TRPV1 antagonist capsazepine (31). Moreover, given the good brain penetration of A-993610, with a plasma:brain ratio near 1 : 1, and its high potency at the TRPV1 receptors (27), it seems likely that trigeminocervical neurons had adequate exposure to the drug. The dose of the drug that was used did show the highest effective antinociceptive potential in models of inflammatory peripheral pain without causing toxic side effects (personal communication P Kym, Abbott Laboratories). Although 16% of trigeminal ganglion cells show TRPV1-like immunoreactivity, and the majority of the cells innervating the dura mater and containing TRPV1 receptors also contain CGRP (32,33), these cells may not be crucially involved in durovascular nociceptive mechanisms. The results here suggest the TRPV1 receptor will not be an important target for the treatment of acute migraine attacks.
TRPV1 receptors have been shown to be present in the periaqueductal grey, hypothalamus, thalamus, amygdala, cortex, trigeminal nucleus caudalis and a number of other regions of the brain in humans, rats and mice (34–37). Various studies have shown activation of these areas via local TRPV1 receptors is possible (38,39). The trigeminocervical complex, periaqeductal grey, hypothalamus and thalamus are all important in signal processing during primary headaches (40), and therefore represent targets for drug development (18). Based on this knowledge the hypothesis was created, that TRPV1 receptors might be a target for migraine treatment.
A-993610 showed no effect on the occurrence and spreading of mechanically induced CSD. This makes this TRPV1 receptor antagonist unlikely to be effective in treatment of aura. Blockade of mechanically induced CSD is seen with topiramate (41) and other migraine preventives also have an effect on CSD (42). Glutamatergic mechanisms are important in CSD and because TRPV1 receptor agonists have been shown to enhance the release of glutamate, TRPV1 receptor antagonists would be expected to reduce glutamate-mediated effects (43–45). Possible explanations for this lack of effect include that CSD initiation and propagation have only a relatively small dependence on pH changes. Four different TRPV1 receptor antagonist profiles and their distinct modes of activation have previously been described (46,47).
One possible confounder of the use of TRPV1 receptor antagonists in vivo is changes in core temperature. During the experiments no consistent changes of body temperature have been seen. Although this may have been predicted as an action of a TRPV1 receptor antagonists, the experiments were conducted with a temperature probe and heating blanket that would have offset any effect. Moreover, any hyperthermia after administration of the drug would have been confounded by the hypothermic reaction to anesthesia.
TRPV1 receptor antagonists have shown convincing effects in animal models of peripheral pain (48,49). Indeed electrophysiological measurements of spinal responses have successfully been used to demonstrate the ability of TRPV1 receptor antagonist to reduce heat-evoked responses (50). Recently, in cat, a small but significant suppression of trigeminocervical transmission elicited by electrical and mechanical stimulation of the dura mater, and receptive testing in the facial area, has been demonstrated by IV administration of the TRPV1 receptor antagonist SB-705498 (51). Interestingly, the authors concluded that the compound (SB-705498) may not be useful in the acute treatment of migraine, the focus of our study, but demonstrated the ability to inhibit or even reverse sensitization. Therefore the process of sensitization might be blocked by TRPV1 antagonists, but blocking sensitization will not necessarily treat the acute pain as we have shown using well-established models that have a high predictive value for the potential use of drugs in the acute treatment of migraine (52,53). Additionally, the pathophysiology of sensitization, which can be found in migraine patients presenting as cutaneous allodynia, is associated with migraine but not present in all patients. We have extended the translational question investigated by Lambert and colleagues (51) by studying other dimensions of the trigeminovascular system and failed to demonstrate any effect on neurogenic dural vasodilation or on mechanically induced CSD.
In summary, we have not demonstrated any significant role for TRPV1 receptor–mediated trigeminal durovascular mechanisms or for transmission of trigeminocervical nociceptive signals in the rat. Moreover, TRPV1 receptor mechanisms are not crucial for the transmission of CSD in the tested model. The data, taken in comparison to other receptor systems that alter these model systems and have a role in migraine, suggest here that TRPV1 receptor antagonism is unlikely to be an effective strategy for the acute treatment of migraine.
Footnotes
Acknowledgements
We thank Phil Kym at Abbott Laboratories for providing the data on characteristics of A-993610 and providing the compound. Oliver Summ has been supported by a fellowship from the European Federation of Neurological Societies.