
Abstract
The triptans are agonists at serotonin(5-HT) 1B/1D receptors; however, they are also active at 5-HT1A and 5-HT1F receptors. We conducted this series of experiments to further elucidate the site of action of naratriptan using a well-established animal model of trigeminovascular stimulation. Following electrical stimulation of the superior sagittal sinus of the cat, single cell responses (n = 83) were recorded in the trigeminal nucleus caudalis. Most cells (91%) also responded to electrical and mechanical stimulation of cutaneous or mucosal facial receptive fields. The micro-iontophoretic application of naratriptan resulted in a significant suppression of the response to sagittal sinus stimulation (response suppressed by 47 ± 4%, P < 0.001). The effect of naratriptan was significantly attenuated by application of either the 5-HT1B/1D receptor antagonist GR-127935 (P < 0.001) or the 5-HT1A antagonist WAY-100635 (P < 0.05). The response of single cells to receptive field stimulation was also suppressed by microiontophoretic application of naratriptan, but by only 20 ± 3%. Intravenous administration of naratriptan resulted in a similar selective suppression of sagittal sinus vs. receptive field responses in trigeminal neurones. These results indicate that naratriptan has a central effect in the trigeminovascular system, selectively inhibiting afferent activity in craniovascular neurones, via both 5-HT1B/1D and 5-HT1A receptors.
Introduction
Background
Migraine is a common, recurrent headache syndrome that affects a significant proportion of the population. The one-year prevalence of migraine is approximately 6% in males and 15% in females, with a lifetime prevalence of 16% in the whole population (1). While migraine undoubtedly causes significant morbidity for affected individuals, its impact can also be measured in terms of adverse effects on the family – such as cancelled social or domestic activities, even separation or divorce (2) – and economic costs to society in the form of medical consultations, pharmaceutical expenses and days lost from work. In one 12-month period (1989–90), the total direct and indirect costs of migraine in Australia was estimated to be at least £A302 million (3), with the annual cost in the United States up to £US66 billion (4).
Migraine has been termed a ‘vascular’ headache, and there is no doubt that stimulation of the intra- and extra-cranial blood vessels in conscious humans is painful (5). The pain of migraine was for many years attributed to vasodilatation (6), and many drugs that are effective in the acute treatment of migraine, such as ergotamine, sumatriptan and naratriptan, constrict cerebral arteries (7–9).
It may be that the vasoconstrictor action of antimigraine drugs is the mechanism of their clinical effect in migraine. However, it has also been shown that microiontophoretic application of antimigraine drugs into the trigeminal nucleus caudalis significantly reduces the evoked responses of cells to electrical stimulation of cranial blood vessels (10, 11), indicating that these drugs may also act by modulating the response of trigeminovascular neurones at a central site.
The ergot alkaloids, which for many years were the most effective specific antimigraine drugs available, are active at a variety of serotonin(5-HT) receptors, including 5-HT1A, 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C and 5-HT3 receptors (12). However, the development of the triptans (first sumatriptan, followed by zolmitriptan, naratriptan, rizatriptan and eletriptan, among others) has focused attention on the role of 5-HT1B and 5-HT1D receptors. The triptans have a high affinity for 5-HT1B/1D receptors with most also having a moderately high affinity for 5-HT1A and/or 5-HT1F receptors (13). We have shown that selective activation of either 5-HT1A receptors (by the experimental compound (+)8-OH-DPAT) or 5-HT1B/1D receptors (by the antimigraine drug alniditan) can modulate the trigeminovascular system at the level of the first central synapse (14).
Aims
The first aim of this present study was to use microiontophoretic application of naratriptan, alone and in combination with selective serotonergic antagonists, to answer the specific question of which 5-HT1 receptors might be involved in the known effect of naratriptan on craniovascular sensory transmission and whether it might act at a central site. We hypothesized that naratriptan would suppress the response of second-order neurones in the trigeminal nucleus caudalis, and that it would act via 5-HT1B/1D and 5-HT1A receptors.
We also tested some cells with intravenous administration of naratriptan. This was done to ascertain the effect of peripheral administration of clinically relevant drug doses, thereby avoiding the problems which may be associated with potentially high local concentrations being achieved by microiontophoresis and thus more closely approximating the use of the drug in migraine patients. We could also then ascertain whether the effect of naratriptan varied with different routes of administration.
The second aim was to determine whether naratriptan, administered by either the microiontophoretic or intravenous route, had a similar differential effect on trigeminovascular and cutaneous receptive field input as we had previously demonstrated for alniditan. We hypothesized that this would be a class effect, that is, that naratriptan would have a selective effect on trigeminovascular nociceptive input.
Methods
Surgery
Experiments were conducted on 37 adult cats (mean mass ± standard deviation (SD) 2.6 ± 0.8 kg). Anaesthesia was induced with halothane (2.5%) followed by an intraperitoneal injection of α-chloralose (60–70 mg/kg), with additional intravenous doses (20 mg/kg) given at regular intervals throughout the experiment. Surgery was conducted under supplementary halothane anaesthesia (0.5–1%). The femoral artery and vein were cannulated to allow continuous monitoring of heart rate and blood pressure, and administration of drugs and fluids, respectively. The trachea was intubated and the animal ventilated with 30% oxygen in air to maintain an end-expiratory CO2 in the range 3.5–4.0%. Core temperature was monitored with a rectal thermometer and maintained at 37–38°C by means of a servo-controlled heating blanket. Throughout the experiment, the animal was immobilized with intermittent doses of intravenous gallamine triethiodide (20 mg). The depth of anaesthesia during immobilization was assessed regularly during the experiment by testing for sympathetic responses to noxious stimulation (pupillary dilatation, tachycardia, increase in blood pressure) and by regularly allowing the effects of gallamine to wear off and testing for withdrawal reflexes by pinching a paw. Supplementary doses of either chloralose or gallamine were given when necessary.
The animal was mounted in a David Kopf stereotaxic frame and the lower brainstem was exposed by a C1 laminectomy and occipital craniotomy. The superior sagittal sinus was exposed and isolated in the following manner: a midline craniotomy (25 mm diameter) was performed and two incisions made in the dura on each side of, and parallel to, the superior sagittal sinus; the superior sagittal sinus and its dural attachments were elevated and a third parallel incision made through the falx. The superior sagittal sinus was draped over a bipolar hook electrode, insulated except for the tips. Current spread to the cortex was prevented by lifting the superior sagittal sinus with the stimulating electrode, passing a plastic sheet through the incision in the falx (between the cerebral cortex and the superior sagittal sinus), and by constructing a well around the craniotomy site and filling it with paraffin oil. The superior sagittal sinus was stimulated with supramaximal square-wave stimulus-isolated shocks (≤150 V, 250 µs duration, 0.3 Hz).
Recording
The central tungsten wire of a multibarrelled electrode (15) was used to record single unit activity in the trigeminal nucleus caudalis. The electrode was placed on the dorsal surface of the brainstem, 1–4 mm caudal to the obex and 2–5 mm lateral to the midline, and advanced to a depth of up to 3300 µm below the surface by means of a piezoelectric microdrive. Single unit activity was amplified, filtered and displayed on an oscilloscope. Peristimulus and poststimulus event histograms (usually 50 ms in length) were compiled from signals passed to a window discriminator (10) and used for postexperiment analysis of the latency and discharge probability of single units.
Receptive fields
Most units responding to electrical stimulation of the sagittal sinus were tested for the presence of a receptive field, which was defined as the region of facial skin or mucous membrane from which a neuronal response could be evoked when stimulated with mechanical or electrical stimuli. Receptive fields were classified as low threshold mechanoreceptive (LTM), nociceptive specific (NS) or wide dynamic range (WDR), according to the response to various stimuli, based on standard criteria (16). Receptive fields were classified as ‘electrical’ if the cell responded only to electrical stimulation. Cells responding only to mechanical stimulation of the cornea were classified as NS. Cells responding to ‘tap’ (17) or intra-oral stimulation were not routinely searched for in these experiments. The centre of each receptive field was stimulated electrically with a pair of stimulus-isolated needle electrodes delivering supramaximal shocks (10–120 V, 250 µs duration, 0.3 Hz).
Analysis of single units and drug effects
Single unit discharges were classified as arising from either cell somata or axons depending on their response to glutamate and the shape of the un-filtered action potential. Somata responded to glutamate with an increase in spontaneous firing rate, or had a biphasic action potential, or both. Only units identified as cell somata were considered for full study, as we expected the site of action of drugs to be on cell somata or the nerve terminals apposing them, rather than on axons.
In every experiment, one barrel of the microelectrode was filled with each of the following: sodium glutamate (1 M), saline (3 M; used as a current-balancing channel) and saline (150 mM; used as a control for drug ejection). Two of the remaining three barrels were filled with naratriptan (N-methyl-3-(1- methy l – 4 – piperidinyl)-1H-indole-5-ethane-sulphonamide, 10 mg/ml). The final barrel contained either the selective 5-HT1B/1D antagonist GR-127935 (2′-methyl-4′-(5-methyl-(1,2,4) oxadiazol-3-yl)-biphenyl-4-carboxylic acid(4-methoxy-3-(4-methyl-piperazin-1-yl)-phenyl)-amide, 10 mg/ml) or the selective 5-HT1A antagonist WAY-100635 (N-(2-(4-(2-methoxyphenyl)-l-piperazinyl) ethyl) – N - (2 -pyridinyl)cyclohexanecarboxamidetrihydrochloride, 5 mg/ml). All drugs were dissolved in water for injection.
Teflon coated silver wires were inserted into each of the 6 electrode barrels and used to deliver current from a Dagan microiontophoresis current generator to the drug and salt solutions. Drugs were ejected with currents of +5 to +150 nA for 2–20 min and retained with currents of − 5 to − 10 nA (opposite polarities for sodium glutamate).
One or more poststimulus histograms, each consisting of 50 consecutive recordings of discharges in response to sagittal sinus or receptive field stimulation, were recorded for each cell under control conditions (in the absence of any drugs), during the ejection of naratriptan, and after naratriptan ejection had ceased. The same sequence, with the same naratriptan ejection currents, was also carried out during the continuous microiontophoretic ejection of antagonists. Ejection of saline (150 mM), with the same current and polarity as used for naratriptan, was used as a control to exclude the possibility of current effect.
The effect of drug ejection on the peripherally evoked response was assessed by calculating the number of discharges per stimulus during drug -ejection as a percentage of the average pre- and post-drug stimulus-induced discharges. We used the ‘critical ratio’ test (a function of the resting spontaneous firing rate and the rate evoked by drug ejection) to classify the response of a cell to microiontophoretically applied compounds (18). A critical ratio of 1.96, equivalent to a 30% change in response, is considered significant at the P < 0.05 level. Responses in the present study were classified as either ‘suppressed’ (reversible reduction in response by ≥30%), ‘no effect’ (<30% change), ‘uncertain’ (if the response reduced by ≥30% during drug ejection but did not return to the predrug level after cessation of drug ejection) and ‘potentiation’ (reversible increase in response by ≥30%). In those cases where cells were tested more than once with a particular drug, the mean response to drug ejection was used when classifying the cell. Responses classified as ‘uncertain’ were not considered in the analysis of drug effects. Where other tests (such as continued response to glutamate or receptive field stimulation) allowed us to confirm that the target neurone had not been lost to recording, the responses of cells to superior sagittal sinus stimulation inhibited by naratriptan but which did not recover were classified as ‘suppressed’ rather than ‘uncertain’.
In some experiments, the effect of intravenous naratriptan was tested on the response of cells to sagittal sinus and receptive field stimulation. Doses of 10–30 µg/kg were used, these being equivalent to the subcutaneous doses used in human studies (19). The responses of cells to both sagittal sinus and receptive field stimulation were recorded before drug injection, then at intervals (5, 10, 15, 20, 30, 45, 60, 75, 90, 120, 150, 180, 210 and 240 minutes) after drug injection. Most cells tested with i.v. naratriptan were first tested with iontophoretic application of naratriptan. Intravenous injection of naratriptan was only made after complete return of control responses to stimulation (10–15 min).
In each case where both sagittal sinus and receptive field evoked responses were present and tested with naratriptan, the shape of the action potential evoked from each stimulus site was recorded to ensure that the same cell was being activated by both sagittal sinus and receptive field stimulation. Action potential morphology was assessed by photographing the unfiltered action potential displayed on the storage oscilloscope and/or by averaging 50 consecutive unfiltered action potentials using a customised averaging program.
Sodium glutamate was purchased from Sigma-Aldrich, Castle Hill, NSW, Australia; naratriptan and GR-127935 were gifts from Glaxo-Wellcome, UK; WAY-100635 was a gift from Wyeth-Ayerst Research, Princeton, NJ, USA.
Histology
Selected recording sites were marked by making an electrolytic lesion with a cathodal DC current (5 µA) passed through the tungsten wire for 5–10 s. At the end of the experiment, the cat was deeply anaesthetized with sodium pentobarbitone (20 mg/kg) and perfused via the aorta with 0.9% saline followed by 10% phosphate-buffered formalin. The lower brainstem/upper cervical spinal cord was removed and stored in phosphate-buffered formalin. The brainstem was later sectioned on a freezing microtome (50 µm sections) and stained with cresyl violet. Recording sites were reconstructed from a combination of electrolytic lesions, track marks and microdrive readings.
Statistical analysis
Data are presented as mean ± SD when population properties are described; when comparisons are made between groups, these are presented as mean ± standard error (SEM). Student's paired two-tailed t-test was used to compare firing rates and the critical ratio test was used to assess changes in the discharge rate of cells.
All experiments described in this paper were approved by the Animal Care and Ethics Committee of the University of New South Wales and conformed to its guidelines.
Results
Response properties of cells
The responses of 83 cell bodies, characterized by their response to electrical stimulation of the superior sagittal sinus, were recorded in the trigeminal nucleus caudalis of 37 cats. These 83 cell bodies were selected from a larger population of single units responding to sagittal sinus stimulation, but which were unsuitable for the purpose of this study either because they were classified as fibres, or because they were lost before baseline or drug testing could be completed. The latency of the initial response of these 83 neurones to sagittal sinus stimulation was consistent with activation of A-δ fibres (mean latency ± SD, 10.4 ± 5.0 ms; estimated conduction velocity 6.6 m/s). Cells responded to electrical stimulation of the sagittal sinus with between 0.5 and 10.1 discharges per stimulus (mean ± SD, 2.6 ± 2.2).
Sixty-four of 71 neurones (91%) tested for a receptive field were found to have one on the ipsilateral face (a receptive field was not found for the remaining 7 cells). Most cells responded to nociceptive stimuli exclusively (NS, n = 12) or preferentially (WDR, n = 33), with only seven responding to innocuous stimuli alone (LTM). Two neurones responded only to electrical stimulation of the receptive field. The response patterns of the remaining 10 were not characterized. The latency of the response of cells to electrical stimulation of the receptive field was 10.2 ± 5.4 ms (n = 64).
Effect of naratriptan on the response to sagittal sinus stimulation
Iontophoretic ejection of naratriptan suppressed the response of neurones to sagittal sinus stimulation by 47 ± 4% (n = 82 trials in 77 neurones), expressed as a percentage of the mean pre- and postnaratriptan controls. This reduction was significant (t = 13.2, P < 0.001, n= 82). In 59 of the 82 trials (72%), neurones were classified as ‘suppressed’ by naratriptan according to the critical ratio test. Suppression of responses commenced within one minute of naratriptan ejection and disappeared over 10–15 min following cessation of ejection.
Microiontophoretic application of saline did not affect the response of cells to electrical stimulation of the sagittal sinus (t = 0.8, P > 0.1, n = 13). These results are summarized in Table 1.
Effect of naratriptan, GR-127935, WAY-100635 and saline on the response of neurones to sagittal sinus stimulation

Cell bodies responding to sagittal sinus stimulation were classified according to their response to microiontophoretic application of individual drugs (critical ratio test).
Effect of naratriptan on the response to receptive field stimulation
In 52 cells tested, microiontophoretic ejection of naratriptan reduced the response to supramaximal stimulation in the centre of the facial receptive field by only 20 ± 3% (n = 77 trials). In only 22 (27%) of these 77 trials was the response to receptive field stimulation classified as ‘suppressed’, according to the critical ratio test. In contrast, the sagittal sinus response of these same cells was classified as ‘suppressed’ by naratriptan in 72% of the trials (mean reduction 49 ± 4%; Table 2). Thus, we saw a selective suppressant effect of naratriptan on sagittal sinus evoked responses. In quantitative terms, the sagittal sinus evoked response was suppressed by an average of 25% more than the receptive field-evoked response was suppressed (t = 4.64, P < 0.001, n= 56). This differential effect of naratriptan is illustrated in Fig. 1.

Poststimulus histograms illustrating the responses of one cell to electrical stimulation of (a,c,e) the superior sagittal sinus and (b,d,f) a cutaneous receptive field in the ophthalmic division of the trigeminal nerve. The three rows of histograms represent responses before (a,b), during (c,d) and after (e,f) microiontophoretic administration of naratriptan. The response to sagittal sinus stimulation was suppressed by 83%, whereas the response to receptive field stimulation was suppressed by only 26%. In these and subsequent poststimulus histograms, the vertical axis represents the total number of discharges recorded for each bin over 50 scans. (Stimulus artefact is seen at the beginning of the x-axis on most histograms).
Effect of naratriptan on the response of the same trigeminal neurones to sagittal sinus and receptive field stimulation

Cell bodies responding to both sagittal sinus and receptive stimulation were classified according to their response to microiontophoretic application of naratriptan (critical ratio test).
Intravenous administration of naratriptan
The effect of intravenous administration of naratriptan (dose range 10–30 µg/kg) on the response to sagittal sinus and receptive field stimulation was assessed in 10 cells. The response to sagittal sinus stimulation of nine of these 10 neurones was classified as ‘suppressed’ by intravenous naratriptan (critical ratio test). The mean response to sagittal sinus stimulation started to decrease five minutes after injection (−13 ± 10%, n = 10, compared to control response) reaching a maximum reduction at a mean of 30 min after naratriptan injection (range 10–60 min). The mean of the maximum reduction over three hours after naratriptan injection was 63 ± 8% (n = 10). This was a significant difference (t = 8.21, P < 0.001).
Eight of the 10 cells tested with intravenous naratriptan were also tested with microiontophoretic application of naratriptan. The responses of seven of these eight cells to sagittal sinus stimulation were suppressed and all of the eight were also suppressed by intravenous naratriptan. The mean suppression by iontophoresis for these 8 cells was 63 ± 8% and for the same cells the mean maximal suppression by intravenously administered naratriptan was 58 ± 7%, both of which were significant (t = 8.21, t = 8.11, P < 0.001, N= 8).
In contrast to the effect on the response to sagittal sinus stimulation, the response of the same 10 cells to receptive field stimulation was suppressed to a much smaller extent. In 6 of the 10 neurones, responses to RF stimulation rose to above baseline levels for up to 30 min, in 2 of them this effect was significant according to the critical ratio test. The mean of the maximum suppression in response to RF stimulation was 21 ± 10% (n = 10). These same cells were suppressed by a mean of 21 ± 10% by iontophoretic application of naratriptan, neither of which was significant (t = 2.21, t = 1.95, n= 10).
The effect of naratriptan is blocked by a 5-HT1B/1D receptor antagonist
Microiontophoretic ejection of GR-127935 alone produced no significant change in the response to sagittal sinus stimulation in 13 cells (mean change in firing 1 ± 11%, t = 0.1, P > 0.1). Microiontophoretic application of naratriptan, in the presence of the continuing ejection of GR-127935, was carried out on 11 neurones responding to stimulation of the sagittal sinus, and in which this evoked response had already been shown to be reversibly suppressed by naratriptan. These results are summarized in Table 3.
Antagonism of the effect of naratriptan by WAY-100635 and GR-127935
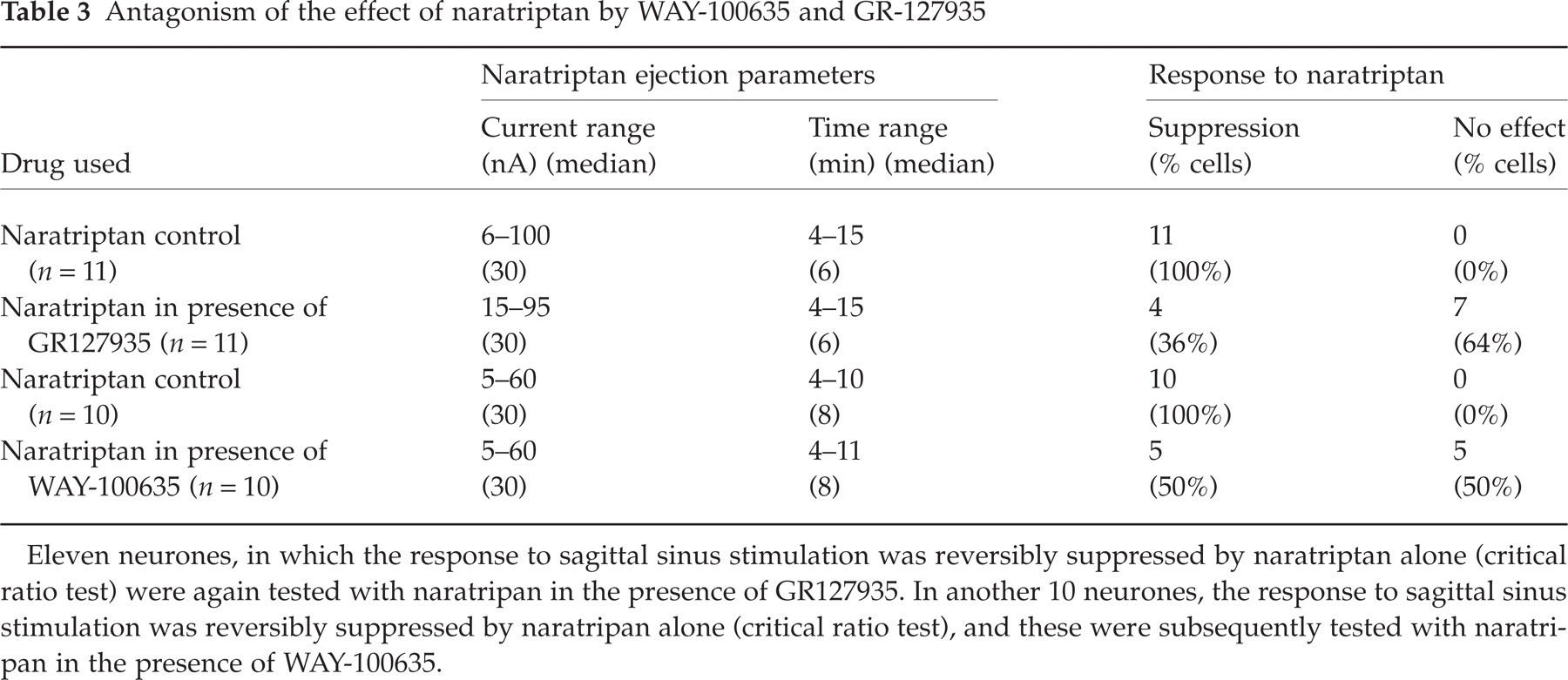
Eleven neurones, in which the response to sagittal sinus stimulation was reversibly suppressed by naratriptan alone (critical ratio test) were again tested with naratripan in the presence of GR127935. In another 10 neurones, the response to sagittal sinus stimulation was reversibly suppressed by naratripan alone (critical ratio test), and these were subsequently tested with naratripan in the presence of WAY-100635.
In seven out of 11 neurones, the classification of the effect of naratriptan on the response to sagittal sinus stimulation was changed from ‘suppression’ to ‘no effect’ in the presence of GR-127935 (critical ratio test). An example is illustrated in Fig. 2. In all four of the remaining neurones, GR-127935 partially antagonized the effect of naratriptan. In the 11 neurones overall, naratriptan alone suppressed the response to sagittal sinus stimulation by 62 ± 5%. In the presence of GR-127935, the same current of naratriptan caused a suppression of only 22 ± 5%. Therefore, GR-127935 reduced the effect of naratriptan by 65% and this represented a significant antagonism of the suppression produced by naratriptan alone (t = 6.1, P < 0.001, n = 11).

The effect of naratriptan and GR-127935 on the response of one cell to sagittal sinus stimulation. In the presence of naratriptan alone, the response was suppressed by 69%. Application of GR-127935 suppressed the response by 22%; in the presence of GR-127935, naratriptan suppressed the response by only 5%.
The effect of naratriptan is partially blocked by 5-HT1A receptor antagonism
Iontophoretic ejection of WAY-100635 alone produced no significant change in the response to sagittal sinus stimulation in nine cells (mean change in firing 1 ± 6%, t = 0.2, P > 0.5).
Microiontophoretic application of naratriptan, in the presence of the continuing ejection of WAY-100635, was carried out on 10 neurones responding to stimulation of the sagittal sinus, and in which this evoked response had already been shown to be reversibly suppressed by naratriptan. These results are summarized in Table 3.
In five out of 10 neurones, the classification of the effect of naratriptan on the response to sagittal sinus stimulation was changed from ‘suppression’ to ‘no effect’ in the presence of WAY-100635 (critical ratio test). In one of the remaining five neurones, WAY-100635 partially antagonized the effect of naratriptan, and in four neurones the effect of naratriptan was unchanged. In the 10 neurones overall, naratriptan alone suppressed the response to sagittal sinus stimulation by 56 ± 4%. In the presence of WAY-100635, the same current of naratriptan caused a suppression of only 27 ± 11%. Therefore, WAY-100635 reduced the effect of naratriptan by 52% and this represented a significant antagonism of the suppression produced by naratriptan alone (t = 2.6, P < 0.05, n= 10).
Histology
The location of 44 cells was determined from histological sections. Fourteen (32%) cells were found in layers I/II, 18 (38%) in layers III/IV and 8 (18%) in layers V/VI of the medullary dorsal horn (20). Four cells were found outside the trigeminal nucleus caudalis: two were close to the edge of the nucleus, but two were in the ventral horn of the medullary grey matter, in the region of the nucleus ambiguus.
Neurones in layers I/II and layer V tended to be more responsive to the effect of naratriptan than neurones in layers III/IV: 71% (layers I/II) and 75% (layer V) were suppressed by naratriptan, vs. 44% in layers III/IV.
Not all recording sites were marked during the course of an experiment, due to concerns that repeated electrolytic lesions might adversely affect the recording characteristics of the electrode. In addition, some lesions were unable to be recovered even after histological processing. However, all cells (whether or not histologically verified) were found within the same range of stereotaxic and microdrive coordinates, responded to sagittal sinus stimulation and, in most cases, facial receptive field stimulation, consistent with their being located in trigeminal nucleus caudalis.
Discussion
Intravenous administration of naratriptan has previously been shown to inhibit the response of neurones in the upper cervical spinal cord of the cat to electrical stimulation of the superior sagittal sinus, an effect that was antagonized by intravenous pretreatment with the selective 5-HT1B/1D antagonist GR-127935 (21). In the present study, we used the technique of microiontophoresis, rather than intravenous administration of drugs, because we were interested in testing the hypothesis of a central site of action by delivering drugs into a localized region of the central nervous system, and in performing multiple pre- and postdrug tests in the one animal. Our previous study (14) was undertaken using two serotonergic agonists: (+)8-OH-DPAT, which is a pharmacological tool, and alniditan, which is an effective antimigraine drug (22) but has not gone into clinical use. We therefore wanted to determine whether microiontophoretic application of naratriptan, an effective (23) and widely available antimigraine drug, would have the same effect on the trigeminovascular system, and to use selective antagonists to determine which 5-HT1 receptors were involved with these effects.
In this present study, we have demonstrated that the inhibitory effect of naratriptan on trigeminovascular neurones was mediated predominantly by 5-HT1B/1D receptors, with a smaller but still significant contribution from 5-HT1A receptors. In addition, naratriptan had a selective effect on trigeminovascular vs. receptive field input into the same trigeminal neurones, and this selectivity was seen for both microiontophoretic and intravenous routes of administration.
The response of 73% of cells to sagittal sinus stimulation was suppressed by microiontophoretic application of naratriptan, and the mean suppression was 48% compared to control values. The responses of some cells were not suppressed, and this may have been due to inadequate drug concentration at the site of action: the evoked potentials of cells may have been recorded from a distance over which the drug was unable to diffuse.
Naratriptan has a high affinity for recombinant human 5-HT1B and 5-HT1D receptors (pKi 0.47 nM and 0.69 nM, respectively) (24) with a relatively lower affinity for 5-HT1A receptors (pKi 26 nM) (25). The mean maximum plasma concentration of naratriptan in humans after an oral 2.5 mg dose is 12.6 ng/ml (26), which is equivalent to 33.9 nM (molecular weight 371.9) (27). Therefore, at clinically relevant doses, the plasma level of naratriptan is sufficient to allow binding to 5-HT1A, 5-HT1B and 5-HT1D receptors.
In an in vitro model of drug efficacy, naratriptan is more effective at recombinant human 5-HT1D and 5-HT1B receptors (EC50 4.4 nM and 23 nM, respectively) (24) than at 5-HT1A receptors (EC50 79 nM) (25). Again, the plasma levels achieved by clinically relevant doses of naratriptan exceeds the EC50 for 5-HT1B and 5-HT1D receptors, and is the same order of magnitude as the EC50 for the 5-HT1A receptor, indicating that a good drug effect is achieved at 5-HT1B and 5-HT1D receptors, with a lesser effect at 5-HT1A receptors.
Application of either the selective 5-HT1B/1D antagonist GR-127935, or the selective 5-HT1A antagonist WAY-100635, significantly reduced the effect of naratriptan. However, some degree of suppression of neuronal firing by naratriptan was still apparent even in the presence of either of these antagonists. This may be explained by the fact that GR-127935 antagonized the effect of naratriptan at 5-HT1B/1D but not 5-HT1A receptors, thereby unmasking the effect of naratriptan at 5-HT1A receptors. Conversely, WAY-100635 antagonized the effect of naratriptan at 5-HT1A, but not 5-HT1B/1D, receptors. It may also be the case that, at times, insufficient concentration of the appropriate antagonist was delivered to the receptors at which naratriptan was acting, thereby underestimating the effect of these antagonists. We did not administer both WAY-100635 and GR-127935 together, for technical reasons: only three of the six electrode barrels were available for agonist/antagonist use, and because of intermittent episodes of the electrode barrels ‘blocking’ and failing to administer drugs, we used naratriptan in two of the three barrels to minimize the risk of failing to administer the agonist. Therefore, only one barrel per experiment was available for antagonist administration.
5-HT1A receptors have been demonstrated to exist in the medullary and/or spinal dorsal horns of the cat (28), rat (29) and human (30). In humans, 5-HT1B (and not 5-HT1D) receptors were found on the middle meningeal artery, whereas 5-HT1D (and not 5-HT1B) receptors were found on the central terminals of trigeminovascular afferent nerves (31), which themselves were in close proximity to second-order neurones in the trigeminal nucleus caudalis.
The pattern of 5-HT1D receptor distribution is one reason to conclude that naratriptan was acting at a presynaptic site in the present study. In addition, the selective effect of naratriptan in suppressing trigeminovascular, but not cutaneous, afferent input in the same second-order trigeminal neurones implies naratriptan was acting presynaptically on the trigeminovascular afferent terminal, perhaps blocking the release of a nociceptive neurotransmitter such as CGRP, with a lesser effect on the presynaptic cutaneous afferent. This may reflect a difference in the density of presynaptic 5-HT1B and/or 5-HT1D receptors on the different afferents, but to our knowledge no study has yet assessed the distribution of serotonin receptors on the central terminals of trigeminovascular vs. cutaneous afferents.
In the present study, intravenous naratriptan (10–30 µg/kg) suppressed the response of nine of 10 trigeminal neurones to sagittal sinus stimulation. Eight cells were tested with both intravenous (mean suppression of response 58%) and microiontophoretic (mean suppression 63%) drug administration. Following intravenous naratriptan, the suppression of responses to sagittal sinus stimulation started five minutes after injection, reached a maximum effect after 30 min, and persisted for over 150 min in some cases. A previous study (21) had also shown that intravenous naratriptan, in doses of 30 and 100 µg/kg, suppressed the response of trigeminal neurones to sagittal sinus stimulation by approximately 57%. These authors also demonstrated an effect of naratriptan lasting at least up to 25 min, although the maximum suppression of trigeminal response was seen after 10 min. In addition, these authors showed that the effect of intravenous naratriptan was antagonized by pretreatment with intravenous GR-127935, again demonstrating the role of 5-HT1B/1D receptors in the effect of naratriptan.
The time course of the onset of the suppression of the response to sagittal sinus stimulation in the present study is similar to the time course of headache relief reported by patients following subcutaneous naratriptan administration (19; Fig. 1 p. 472). Patients reported headache slowly improving from 10 min post injection, and all groups showed a significant difference from placebo by 60 min. Maximum headache relief was reported by 90 min, and the effect persisted for at least 120 min, in some cases up to 24 h.
As with microiontophoretic administration, a differential effect on neuronal responses to sagittal sinus and receptive field stimulation was also noted following intravenous administration of naratriptan. However, in the latter instance the response to receptive field stimulation sometimes showed a slight increase, rather than being immediately suppressed, indicating that naratriptan was possibly sensitizing peripheral cutaneous nerves or receptors to stimulation. It is unlikely that the augmentation of response to receptive field stimulation with some neurones was a central effect, as it did not occur following microiontophoretic administration of the drug, where the reverse effect was seen. This result is interesting, as sensitization of cutaneous receptors may explain the paraesthesia reported by patients taking naratriptan (19, 23).
Other authors (25) have noted the significant affinity of drugs used in the acute treatment of migraine (including dihydroergotamine, sumatriptan and naratriptan) for recombinant human 5-HT1A receptors, and their efficacy at these receptors in an in vitro model. Like these authors, we do not propose that 5-HT1A receptor agonism is the primary mode of action of these drugs, but rather an adjunct to their clinical effect. In a previous study (14), we found that the evoked response of trigeminovascular neurones was significantly suppressed by the selective 5-HT1A agonist (+)8-OH-DPAT. However, the effect of this compound was quantitatively less than the effect of alniditan at 5-HT1B/1D receptors. In the present study, we found that 5-HT1A receptor activation by the antimigraine drug naratriptan also suppressed nociceptive trigeminovascular transmission; others have questioned whether 5-HT1A receptor activation may alleviate nausea and vomiting (32). There is some clinical evidence, from an open trial, that the oral 5-HT1A agonist buspirone may reduce the frequency and severity of migraine (33).
The dose of intravenous naratriptan used in this study was equivalent to the parenteral doses used, and found to be efficacious, in human subjects (19). When drugs are administered by microiontophoresis, it is difficult to be certain of the exact amount of drug delivered, although local drug concentrations near the electrode tip are likely to be high. The finding that intravenous naratriptan had a similar effect on trigeminal neurones to that achieved by microiontophoretic administration of the drug is further evidence that the systemically administered drug has a central effect. It also validates the use of microiontophoretic drug administration as a means of testing potential antimigraine drugs, particularly when looking for an effect at central sites.
In this study, we have looked at the effect of naratriptan at one central site – the trigeminal nucleus caudalis. However, evidence is accumulating in humans that other brainstem sites such as the periaqueductal grey matter may be the sites of origin of migraine itself (34–36). In addition, it has been demonstrated that intravenous administration of naratriptan in the rat modulates the spontaneous activity of neurones in the nucleus raphe magnus, thereby suggesting an effect on the endogenous pain control system (37). Thus, it is becoming apparent that not only does the brainstem play an important role in migraine, but also that the antimigraine drugs may well act at several different levels within the central nervous system to modulate nociceptive transmission.
Despite the remarkable clinical effect of the triptans in the treatment of migraine, there remain a significant proportion of patients who do not benefit from their use, either because the drugs fail to relieve their headache, or the headache recurs, or they are intolerant of the medication (38). Although further improvement in receptor specificity may be desirable, the results of this study indicate that, in addition to 5-HT1B/1D receptors, 5-HT1A receptors may also play a role in the clinical effect of naratriptan and other acute antimigraine agents.
Footnotes
Acknowledgements
We would like to acknowledge the technical assistance of Mr George Mallos. This research was supported by grants from the National Health and Medical Research Council of Australia and the Australian Brain Foundation. Dr Boers was supported by a National Health and Medical Research Council Postgraduate Medical Research Scholarship.