
Abstract
Central sensitisation is a key mechanism of migraine; understanding its modulation by anti-migraine drugs is essential for rationalising treatment. We used an animal model of central trigeminal sensitisation to investigate neuronal responses to dural electrical stimulation as a putative electrophysiological marker of sensitisation and its modulation by ketorolac. In anaesthetised rats, responses of single convergent wide-dynamic range neurons of the spinal trigeminal nucleus to dural electrical simulation were recorded in parallel to their ongoing activity and responses to facial mechanical stimulation before and after a short-term dural application of an IS. Both ongoing activity and responses to dural electrical stimuli were enhanced by the inflammatory challenge, whereas neuronal thresholds to mechanical skin stimulation were reduced (p < .05, N = 12). Intravenous ketorolac (2 mg/kg, N = 6) reduced ongoing activity and responses to dural electrical stimulation, and increased mechanical thresholds versus vehicle controls (p < .05, N = 6). We conclude that neuronal responses to dural electrical stimulation can serve as a suitable marker which together with admitted electrophysiological signs can objectively detect central trigeminal sensitisation and its modulation by anti-migraine treatments in this preclinical model of migraine.
Introduction
The pathogenesis of migraine attacks is believed to involve trigeminovascular events, leading to the vicious circle of activation of nociceptive afferents, neurogenic inflammation of the dura and central sensitisation (1). As the relative contribution of these mechanisms may change over the course of the attack, understanding the mechanism of action of anti-migraine drugs is essential for rationalising the treatment to achieve maximum efficacy.
Central sensitisation is a key mechanism responsible for headache, allodynia and hyperalgesia, and potentially leads to chronic headaches (2–4). It is triggered by prolonged nociceptive input from the dura and involves hyperexcitability of second- (and potentially higher-) order neurons mediating nociceptive transmission from intracranial vasculature to higher centres in the central nervous system. Targeting central sensitisation with anti-migraine treatments is essential for maximising the therapeutic benefit and preventing the risk of chronification (5,6). Therefore, preclinical models that involve central sensitisation can be useful for investigating new anti-migraine treatments as well as confirming mechanisms of action of established drugs.
Electrophysiological experiments have shown that dural sensitisation by inflammatory mediators (e.g. application of an ‘inflammatory soup’) leads to long-term increase of background activity of trigeminal second-order neurons and to decrease in nociceptive threshold and expansion of their receptive fields—that is, the hallmarks of central sensitisation (7). This model has been used to study the mechanisms of action of several anti-migraine drugs in order to understand some features of these drugs as seen in the clinical setting. Thus, the 5HT1B/1D receptor agonist sumatriptan has been shown to operate mainly at the peripheral level and be unable to reverse established sensitisation of spinal trigeminal neurons (8,9). This fits with the clinical observations that triptans may be ineffective when used to stop a fully developed rather than early migraine attack in some patients. On the other hand, non-steroidal anti-inflammatory drugs (NSAIDs) such as ketorolac, indomethacin and naproxen can be used successfully to alleviate headache during developed migraine attacks; this has been explained by suppression of central sensitisation (6,10).
In the 1990s, evidence of an attenuating effect of triptans on responses of the spinal trigeminal neurons to superior sagittal sinus electrical stimulation was obtained (11–13). It should be noted that these data were gained in a model without sensitisation of central neurons. As far as electrical stimulation is a convenient, well-standardised way to cause nociceptive effect (14), it can be easily integrated with pre-clinical screening and allows the detection of a drug's action with proposed or clinically proved anti-migraine activity on nociceptive transmission in trigeminovascular system. However, the experimental model without preliminary sensitisation does not fully mimic the pathological processes developing during migraine attack and does not allow profound study of the pharmacodynamics of a substance (9).
At the same time, in the model with sensitisation, dural electrical stimulation was used only for identification of spinal trigeminal neurons, whereas for monitoring of evoked neuronal activity dural mechanical stimulation was applied (6,7,9,10). Thus, neither electrically induced activity of sensitised trigeminal neurons or its modulation by anti-migraine drugs have been studied. Meanwhile, analysis of changes in dural-induced electrical responses of the spinal trigeminal neurons might give us valuable information about the dynamics of their functional status during sensitisation and after treatment. In our opinion, the electrical responses might serve as additional marker of central sensitisation, which in aggregate with other admitted neurophysiological signs, allows maximum objective estimation of neuronal excitability at different stages of an experiment.
Therefore, in the present work we investigated this putative marker in the animal model of trigeminal pro-nociceptive sensitisation evoked by dural inflammation. We studied responses of sensitised convergent trigeminal neurons to dural electrical stimulation in parallel to their responses to mechanical stimulation of the face skin and their background activity. In addition, we investigated the effects of a typical NSAID, ketorolac, on these markers of sensitisation of central trigeminal neurons.
Materials and methods
General procedures
All experiments adhered to the current Internaional Association for the Study of Pain (IASP) guidelines and were approved by the Research Ethics Committee of Saint-Petersburg State Medical University. Twelve Wistar male rats (body weight 300–390 g) were anaesthetised with urethane (800 mg/kg, intraperitoneally [IP]) and α-chloralose (60 mg/kg, IP). Catheters were placed into the femoral vein for administration of anaesthetics and test drugs, and into the femoral artery for continuous monitoring of blood pressure. The trachea was intubated and the head of the animal was fixed in a stereotaxic frame. The neck muscles overlying the cisternа magna were separated along the midline and C1 laminectomy was performed. The dura mater was removed to expose the medulla and C1 spinal cord. A longitudinal craniotomy close to the superior sagittal sinus was performed and stimulating electrodes were placed on the dura mater. The animal was paralysed with pipecuronium bromide (intravenous [IV], 1.2 mg/kg initially, maintenance 0.6 mg/kg as required) and artificially ventilated with room air (75–100 cycles/min, 2–4 ml/cycle) using a small-animal ventilator. Rectal temperature was maintained between 37–38°C by means of a servo-controlled heating pad. The depth of anaesthesia was assessed by monitoring blood pressure responses to noxious stimulation; supplementary anaesthetic was administered when necessary to ensure the absence of gross (>20% from baseline level) blood pressure fluctuations.
Electrical stimulation of the dura mater
Bipolar stimulating electrodes had resistance of 50 KΩ and consisted of two varnish-insulated silver wires with beads (0.3 mm in diameter) at the end. The dura mater was stimulated with single rectangular pulses of 500–1000 µA (25–50 V) at a duration of 0.8 ms (one stimulus per 3 s) delivered by a computer-controlled stimulator connected to a stimulus isolation unit. The stimulus intensity was 1.5 times the response threshold.
Extracellular recordings
Neuronal activity was recorded by varnish-insulated tungsten microelectrodes (Science Products GmbH, Hofheim, Germany) with a tip diameter of 5 µm and a resistance of 12 MΩ. The electrodes were lowered into the spinal trigeminal nucleus at the level of the C1 spinal cord in 4-µm steps using a micro-drive. Recording sites were limited to a region from 0.0–3.0 mm caudal to the obex and from 1.5–3.5 mm left to the middle line, at a depth of 0.2–1.3 mm relative to the dorsal surface of the spinal cord ipsilateral to the stimulation site. Signals from the recording electrode were amplified and passed to the analogue input of an A/D converter of an IBM-compatible computer by means of a multifunctional acquisition card (sampling period 25 µs). For online acquisition, processing and displaying of data, custom written software was used. To isolate activity of single units from adjacent cell potentials and noise, three-level amplitude discrimination was used on-line. Signals of low-level amplitude were rejected as noise. Action potentials with amplitude at middle and maximal levels were considered as generated by individual neurons and processed separately. Ongoing activity of trigeminal neurons and their responses to electrical stimulation of the dura mater were analysed as peristimulus time histograms, such that signals gated though the amplitude discrimination were collected in successive bins of 1 ms. For evoked responses, data were collected from 20 recordings (one per 3 s) over 50 ms after each electrical stimulus. For histograms of ongoing activity, pseudostimulation was used, that is, the same software as for creating histograms of evoked responses was used but electrical stimulation was not really applied. The histograms had a sweep length of 500 ms (250 ms before/after the pseudostimulus onset) and were created automatically from 100 recordings (one per 1 s). All recorded units apart from responses to electrical stimulation of the dura mater were tested for responses to von Frey mechanical stimulation of the dural and cutaneous receptive fields. Only neurons that demonstrated all three kinds of responses were selected for further testing. For cutaneous stimulation, the thresholds were determined using the five steps up-and-down method with calibrated von Frey filaments (North Coast Medical, Morgan Hill, CA, USA). Filaments were applied to the most sensitive site of the receptive field in ascending and descending order consecutively. The final mechanical threshold was determined as the 5th root taken from multiplication of values obtained in ascending and descending steps.
Experimental protocol
Activation and sensitisation of meningeal nociceptors was induced by dural application of an inflammatory soup (IS) containing 0.5% histamine, 0.3% serotonin, 0.03% prostaglandin E2, 0.3% bradykinin and 0.5% capsaicin at pH 5.5. The recipe was developed empirically. Addition of capsaicin to standard components of IS (6,7) allowed us to considerably potentiate the sensitising effect of the IS. This is the only combination which resulted in a sufficient level of central sensitisation in our preliminary experiments. The exposed dura mater was bathed in IS for 5 minutes followed by a wash with saline. Neurons were selected for further testing only if, within 60 minutes after the IS application, they showed at least two of three signs of sensitisation, such as increase in ongoing activity, enhancement of responses to electrical stimulation of the dura mater and decrease in thresholds to mechanical stimulation of cutaneous receptive fields. Effects of ketorolac (2 mg/kg in 0.6 mL infused IV over 1 minute) or its vehicle (isotonic saline) were tested in two groups of animals (N = 6 each) in 60 minutes after IS application. Neuronal activity was then studied over 60 minutes after ketorolac or vehicle administration. Recordings of ongoing and electrically evoked neuronal activity with simultaneous creation of peristimulus histograms were performed before (0 min), and in 15, 30, 45 and 60 minutes after the IS application on the dura mater. If sensitisation of trigeminal neurons was observed, ketorolac or saline was given, and further recordings were performed in 15, 30, 45 and 60 minutes after the administration. In all experiments, only one unit was tested in each animal. At the end of the experiment rats were killed by an overdose of urethane (>3 g/kg, IV). The recording sites within the spinal cord were marked by an electrolytic lesion through the recording electrode. After routine histological processing of the tissue, lesion sites were examined under a light microscope.
Statistical analysis
Using peristimulus histograms, neuronal ongoing activity and electrically evoked responses were expressed as mean number of spikes per second. To estimate changes in neuronal activity after the treatment with ketorolac or saline, the ongoing firing and evoked responses at various time points were normalised and expressed as percentages of the mean value prior to the administration. Raw data were used for analysis of responses to mechanical stimulation of cutaneous receptive fields. Non-parametric Friedman, Wilcoxon and Mann-Whitney tests were used to determine the significance of changes in neuronal activity following the IS application and IV infusion of ketorolac or its vehicle. Statistical significance was set at p < .05. Data are expressed as mean value ± standard error of the mean (SEM). The analysis was carried out using Origin 7.5 (OriginLab, Northampton, MA, USA) and GraphPad InStat 3.02 (GraphPad Software, La Jolla, CA, USA) software packages.
Results
General properties of neurons
Extracellular recordings were made from 12 neurons within the caudal part of the spinal trigeminal nucleus; all of them received convergent afferent inputs from the dura mater and facial skin. All recorded neurons showed low initial ongoing activity (1–6 spikes/s). The mean firing rates of the vehicle- (N = 6) and ketorolac-treated groups (N = 6) did not significantly differ, at 2.2 ± 1.3 spikes/s and 3.8 ± 1.4 spikes/s, respectively.
Neurons of both experimental groups showed an excitatory response to electrical stimulation of the dura mater, usually consisting of two components with latencies corresponding to activation of A-δ and C-fibres (Figure 1A). The number of spikes in the response varied among neurons within each experimental group. At baseline, the mean rates of evoked firing were not significantly different between the groups (vehicle-treated group, 17.6 ± 1.6 spikes/s; ketorolac-treated group, 20.6 ± 3.0 spikes/s).
Representative oscillographic recordings of convergent spinal trigeminal neurons. (A) Response to electrical stimulation of the dura mater. The arrow indicates the time of a single electrical stimulus. (B) Responses to mechanical stimulation of the facial cutaneous receptive field with von Frey filaments. The force of each filament is indicated in grams. (C) Response to dural application of the inflammatory soup (IS). Horizontal lines below are the exposition time (IS) and time scale (20 s). s = seconds. ms = milliseconds.
The recorded neurons had predominantly facial cutaneous receptive fields, corresponding to the ophthalmic division of the trigeminal nerve. In response to cutaneous stimulation with von Frey filaments, neurons of both experimental groups generated pronounced bursts of spikes (Figure 1B). The mean mechanical thresholds did not significantly differ between the vehicle- and ketorolac-treated groups and were 14.2 ± 3.4 g and 16.5 ± 3.9 g, respectively.
Effects of dural application of inflammatory soup
Application of IS on the dura produced an immediate increase in ongoing activity of all recorded neurons (Figure 1C), which remained heightened after removing the IS. Within 60 minutes after application, the mean firing rate of neurons in the vehicle-treated group significantly (p < .01, N = 6) increased to 8.1 ± 0.8 spikes/s (Figure 2A). Similarly, in the ketorolac-treated group, ongoing activity significantly (p < .01, N = 6) rose to 10.1 ± 0.8 spikes/s.
Summary diagrams demonstrating the effects of dural application of the inflammatory soup and of intravenous (IV) infusions of saline and ketorolac on ongoing activity of spinal trigeminal neurons. (A) Mean firing rates (in spikes/second) of neurons in the saline- (solid bars) and ketorolac-treated groups (hatched bars). Dashed lines indicate time of inflammatory soup (IS) application and infusion of saline or ketorolac. Time intervals in minutes: 0, initial (baseline) level; 15–60, the first hour; and 75–120, the second hour after IS application. The data are shown as mean number of spikes/second in a given time interval ± standard error of the mean. Significant differences are indicated as follows: **Significantly different from the initial level (p < .01). #Significantly different (p < .01) from the sensitisation level (60 min after end of IS application, just prior to saline or ketorolac infusion). (B) Normalised ongoing activity of the same neurons expressed as percentages of the sensitisation level. Time 0 indicates IV infusion of saline or ketorolac. *Significant differences between the ketorolac and vehicle groups (p < .05). s = seconds. ket = ketoralac. veh = vehicle.
After application of IS, both groups of neurons showed enhanced responses to electrical stimulation of the dura mater (Figure 3A, Figure 4). Within 60 minutes after IS application, the evoked neuronal firing in the vehicle-treated group increased to 31.7 ± 2.3 impulse/s. Similarly, neurons of the ketorolac-treated group demonstrated a significant (p < .01, N = 6) increase in electrically induced responses to 32.8 ± 0.7 impulse/s.
Summary diagrams showing the effects of dural application of the inflammatory soup (IS) and of intravenous (IV) infusions of saline and ketorolac on responses of spinal trigeminal neurons to electrical stimulation of the dura. (A) Mean firing rates (in spikes/second) of neurons in the saline- (solid bars) and ketorolac-treated groups (hatched bars). Dashed lines indicate time of IS application and infusion of saline or ketorolac. Time intervals in minutes: 0, initial (baseline) level; 15–60, the first hour; and 75–120, the second hour after IS application. Data are shown as mean number of spikes/second in a given time interval ± the standard error of the mean. Significant differences are indicated as follows: Significantly different from the initial level (*p < .05, **p < .01); #significantly different (p < .01) from the sensitisation level (60 min after end of IS application, just prior to saline or ketorolac infusion). (B) Normalised evoked activity of the same neurons expressed as percentages of the sensitisation level. Time 0 indicates IV infusion of saline or ketorolac. **Significant differences between the ketorolac and vehicle groups (p < .01). s = seconds. ket = ketoralac. veh = vehicle. Representative histograms showing the effects of pro-inflammatory dural sensitisation and of intravenous ketorolac on neuronal responses to dural electrical stimulation. Activity of the same neuron is shown before (top), 60 minutes after inflammatory soup application (middle) and 60 minutes after ketorolac infusion (bottom). Horizontal axis shows time in milliseconds; vertical axis shows the number of spikes/ms bin. The histograms are produced from 20 stimuli each. ms = millisecond.

The dural sensitisation was accompanied by an increase of neuronal response to mechanical stimulation of cutaneous receptive fields (Figure 5). Thus, in the vehicle-treated group, the mean von Frey threshold decreased to 12.9 ± 4.9 g at 30 minutes and to 7.2 ± 1.6 g at 60 minutes following IS application. Neurons of the ketorolac-treated group showed similar, but more pronounced, changes in response to cutaneous mechanical stimulation. In 30 minutes after IS application, their mechanical thresholds decreased to 9.0 ± 1.1 g, and in 60 minutes this threshold was 8.0 ± 2.5 g. In both groups, in 60 minutes after IS application the mean thresholds to cutaneous mechanical stimulation were significantly different (p < .05, N = 6) from baseline levels (Figure 5).
Pooled data demonstrating changes of neuronal thresholds to mechanical stimulation of facial skin following dural application of inflammatory soup (IS) and intravenous infusion of saline or ketorolac. Solid bars indicate the vehicle- and hatched bars ketorolac-treated groups. Dashed lines show the time of IS application and infusion of saline or ketorolac. Time 0 indicates initial threshold prior to the soup application. The thresholds are shown as mean force of von Frey filaments in grams ± standard error of the mean. Significant differences are indicated as follows: #Significantly different from the initial level (p < .05). *(p < .05) and **(p < .01) significantly different from the vehicle group. Significantly different from the threshold +(p < .05) before ketorolac administration (60 min). ket = ketoralac. veh = vehicle.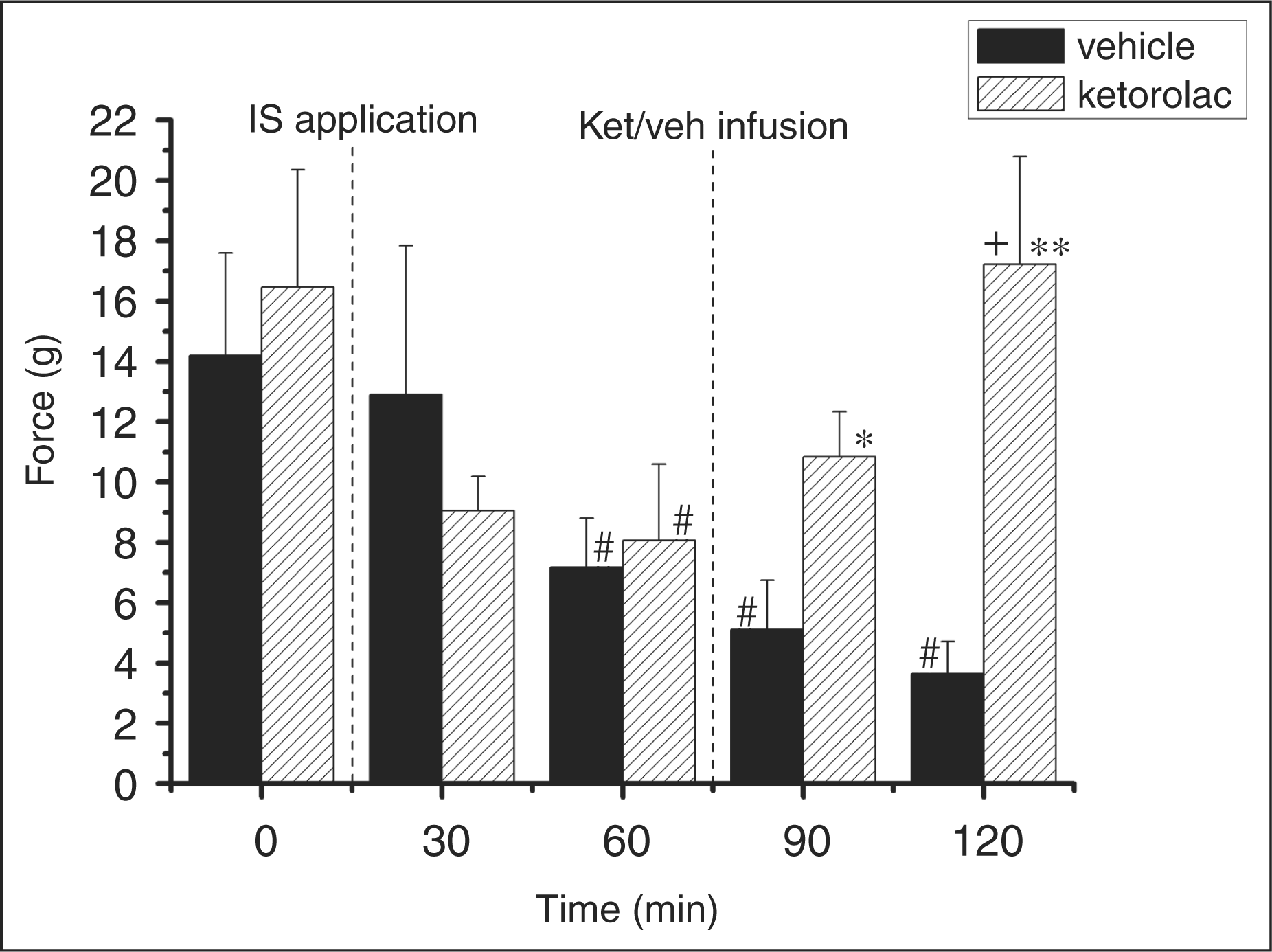
Thus, by the time of IV administration of saline or ketorolac (60 min after IS application onto the dura), neurons of both experimental groups showed at least two of the three signs of central sensitisation: increase in ongoing activity, enhancement of response to electrical stimulation of the dura mater and decrease in thresholds to mechanical stimulation of cutaneous receptive fields.
Effects of systemic administration of ketorolac
Ongoing activity. Administration of ketorolac resulted in gradual inhibition of ongoing activity of sensitised trigeminal neurons. Fifteen minutes after administration, the mean firing rate significantly (p < .05, N = 6) decreased to 58 ± 14% and in 60 minutes to 38 ± 16% of the level before ketorolac infusion (Figure 2B). In total, the mean firing rate within 75–120 minutes significantly (p < .01, N = 6) decreased in comparison to the value prior to the ketorolac administration, to a level (3.6 ± 0.4 spikes/s) that was not different from the baseline activity (0 min) before IS application (Figure 2A). In contrast, ongoing activity of neurons in the vehicle-treated group continued to increase. Thirty minutes after saline administration, it was maximal at 150 ± 33% of the value prior to administration (Figure 2B). Activity then gradually declined and by 60 minutes was 78 ± 39% of the sensitised (pre-infusion) level. As a whole, within 75–120 minutes the mean rate of ongoing activity significantly (p < .01, N = 6) increased in comparison to the initial level (0 min); this level (10.2 ± 0.9 spikes/s) was not different from the value prior to saline administration (Figure 2A). Between-group comparison revealed that the decrease in ongoing activity of ketorolac-treated neurons was significant compared to the vehicle-treated ones (Figure 2B).
Responses to electrical stimulation of the dura. Electrically evoked responses of the studied trigeminal neurons, which were increased after IS application, substantially declined following ketorolac administration (Figure 3A, Figure 4). The decrease in the mean rate of evoked firing was noticeable at 15 minutes after ketorolac administration, and at 60 minutes it was 63 ± 4% of the value prior to administration (Figure 3B). Within the period of 75–120 minutes, electrically evoked activity fell to 22.1 ± 0.9 spikes/s; this level was comparable to the initial level but significantly (p < .01, N = 6) lower than the value before ketorolac administration (Figure 3A). In the group that received IV saline, the mean rate of evoked neuronal activity 15 minutes after the infusion was 142 ± 13% of that prior to saline infusion(Figure 3B). Within the next 45 minutes it gradually decreased, and at the end of recording was 113 ± 13% of the value before saline infusion. From 75–120 minutes after IS application, the mean rate of electrically evoked firing was significantly enhanced (39.1 ± 2.9 spikes/s, p < .05, N = 6) in comparison to the initial level (Figure 3A), which did not significantly differ from the value before saline administration. The inhibition of electrically evoked responses observed in the ketorolac-treated group was significant (p < .01) in comparison to the vehicle-treated group at all time points after saline or ketorolac administration (Figure 3B).
Response thresholds to mechanical stimulation of the skin. Thresholds of neuronal responses to mechanical stimulation of cutaneous receptive fields, which were lowered after dural application of IS, were increased in the group treated with ketorolac (Figure 5). In 60 minutes after ketorolac administration, the mean von Frey threshold increased to 17.2 ± 3.5 g; this value was significantly higher than that prior to ketorolac administration (p < .05, N = 6) and comparable to the initial threshold before inflammatory sensitisation of the dura mater. IV administration of saline in the vehicle-treated group did not affect the IS-induced decrease in thresholds of neuronal responses to mechanical simulation. The mean von Frey threshold continued to decrease over time, and by 60 minutes after the saline administration it averaged 3.6 ± 1.1 g. The reduction of mechanical sensitivity of facial receptive fields observed in the ketorolac-treated group was significant in comparison with the vehicle control group at 30 minutes (p < .05) and 60 minutes (P < .01) after infusion (Figure 5).
Thus, IV administration of ketorolac significantly reduced ongoing activity of the studied trigeminal neurons as well as their responses to electrical stimulation of the dura mater sensitised by dural application of IS. Furthermore, neurons treated with ketorolac showed an increase in von Frey thresholds to mechanical stimulation of cutaneous receptive fields, reversing their sensitivity to pre-sensitisation levels. IV saline had little effect on spontaneous or evoked neuronal activity, which remained elevated compared to pre-sensitisation levels.
Discussion
Consistent with previous reports (7,15), this study demonstrates that short-term activation of dural nociceptors by pro-inflammatory agents results in central sensitisation at the level of trigeminal second-order neurons. This sensitisation manifests not only as long-lasting increase in ongoing activity of these neurons and decrease in their thresholds to stimulation of cutaneous receptive fields, as described previously, but also as enhancement of neuronal responses to electrical stimulation of the dura.
Enhanced ongoing activity of the spinal trigeminal neurons is hypothesised to be a neurophysiological correlate of headache in migraine patients, and its levels may be related to the intensity of pain sensation (9). The increase in ongoing activity observed during IS application is a typical characteristic of wide dynamic range convergent trigeminal neurons (15) and can be explained by afferent input from chemically stimulated dural receptors. However, ongoing activity of the spinal trigeminal neurons recorded in the present study not only maintained heightened after removing IS, but also continued to rise. This may be explained by the phenomenon of central sensitisation developing as a result of the brief stimulation of dural afferents by IS. It may also be a result of continued afferent input due to neurogenic inflammation of the dura following initial IS application (16). Such inflammation is known to involve release of vasoactive peptides in the dura (1), resulting in increased meningeal blood flow and continued nociceptor activation. Thus, ongoing activity may not be a definitive and specific marker of central sensitisation.
Another marker of central sensitisation in trigeminal nociceptive pathways is sensitivity of convergent neurons of the spinal trigeminal nucleus to cutaneous input. The clinical correlate of this phenomenon is considered to be the fronto-orbital cutaneous allodynia developing in many migraine patients during the attack (2,9). Consistent with published observations of enhanced responses of convergent trigeminal neurons to mechanical stimulation of experimentally intact facial receptive fields, we observed decreased thresholds to von Frey stimulation of the skin after dural application of IS.
The present study is the first one demonstrating that responses of spinal trigeminal neurons to noxious electrical stimulation of the dura can serve as a marker of central sensitisation. Previous studies (6,7,9) demonstrated increased neuronal responses to innocuous mechanical stimulation of the dura mater contributing to the development of intracranial allodynia. However, these responses are supposed to be sensitive to changes in sensitivity of dural receptors caused by inflammatory agents. On the other hand, noxious electrical stimulation of the dura will directly activate sensory fibres, bypassing the peripheral receptor; thus, changes in neuronal activity evoked by such stimuli can be attributed to changes in central sensitisation with greater certainty. Moreover, in line with the observed changes in ongoing activity, the magnitude of electrically induced neuronal responses in our study maintained increased and continued to rise for at least an hour after removing IS from the dura mater. This also suggests activation of central mechanisms. Sensitisation of second-order trigeminal neurons, once established, has been shown to be independent from peripheral input (7). It is therefore likely that the balance of central versus peripheral pro-nociceptive mechanisms changes during the period following IS application. The initial activation of dural afferents by IS appears sufficient to trigger central plasticity. As this takes place, the previous sub-threshold or threshold nociceptive stimuli of invariable intensity are perceived by sensitised structures of the trigeminal complex as supra-threshold or 'hyperalgesic' ones (4). This phenomenon may underlie the gradual increase in headache intensity occurring during attacks of migraine. With time, peripheral input may decrease as central sensitisation becomes self-maintained. Thus, the increase in electrically induced activity of trigeminal neurons observed in our study after IS application appears to depend on central mechanisms more than on changes in peripheral susceptibility.
In this study the nonselective COX inhibitor ketorolac was used to validate the various markers of trigeminal sensitisation and demonstrated to inhibit them in parallel. Thus, the depression of ongoing activity of sensitised trigeminal neurons was observed over the same period as the reversal of the inflammation-induced facilitation of their responses to dural electrical stimulation back to frequencies of firing comparable to the initial level before sensitisation. At the same time, the thresholds of neuronal responses to mechanical simulation of the neurons' cutaneous receptive field, lowered by dural inflammatory sensitisation, also returned to the initial values. Meanwhile, the infusion of saline in the control group of animals was not followed by regress in signs of sensitisation, which continued to rise up to the end of an experiment. The fact that all three markers of central sensitisation used in this study changed within the same time period following ketorolac administration and had similar time profiles suggests similar underlying mechanisms, thus providing further validation.
It is known that ketorolac, as a typical NSAID possesses both anti-inflammatory and analgesic effects. Taking into account that development of migraine involves aseptic inflammation of meningeal blood vessels and release of prostaglandins, the use of such drugs for stopping headache attacks seems pathogenetically justified (17). Moreover, our data in conjunction with previous reports (6,10,17), indicate that, in addition to peripheral anti-inflamatory effects, ketorolac and other NSAIDs have a direct analgesic action which is unlikely to be only at the level of peripheral afferent endings of the trigeminal nerve. Our data suggest that the mechanisms of ketorolac action involve reversal of hyperexcitability of spinal trigeminal neurons which, in turn, will result in inhibition of nociceptive traffic to the higher nervous structures and in decrease (normalisation) of peripheral sensitivity. In migraine patients it appears as reduction of pain sensation and decrease in signs of cutaneous allodynia after using ketorolac.
How is the direct inhibitory effect of ketorolac on hyperexcitable spinal trigeminal neurons mediated? This cannot be inferred from this study, but at least three mechanisms can be hypothesised. First, pathologically enhanced activity of central neurons may lead to increased expression of COX1 and COX2, both of which are inhibited by ketorolac. This has been shown to occur at the spinal level in models of somatic pain (18–20). Second, COX inhibitors such as ketorolac may enhance descending antinociceptive influence on neurons within the spinal dorsal horn, a homolog of which is the spinal trigeminal nucleus (21). And finally, ketorolac, as well as other NSAIDs, may have a direct membrano-stabilising effect resulting in modulation of ion channels and recovery of the proper level of the neuronal membrane polarisation (22,23). These hypotheses will need to be confirmed or refuted in future studies.
In conclusion, the present study demonstrated that central sensitisation developing following a brief inflammatory challenge of the dura mater can be reliably detected by assessing ongoing activity of spinal trigeminal neurons, their thresholds to cutaneous mechanical stimuli and responses to electrical stimulation of the dura. These markers are also sensitive to pharmacological interventions able to reverse central sensitisation, such as NSAIDs. The time course of changes in these markers in responses to inflammatory stimulation of the dura or following ketorolac appears similar, suggesting similar underlying mechanisms. This allows us to consider responses of trigeminal neurons to dural electrical stimulation as a suitable marker of central sensitisation, which together with admitted electrophysiological signs such as their ongoing activity and responses to cutaneous mechanical stimulation, can objectively detect neuronal excitability and its modulation by anti-migraine treatments.
Footnotes
Acknowledgements
This study was supported by GlaxoSmithKline.