
Abstract
Objectives: This is the first of a pair of studies designed to assess efficacy, safety and tolerability of onabotulinumtoxinA (BOTOX®) as headache prophylaxis in adults with chronic migraine.
Methods: The Phase III REsearch Evaluating Migraine Prophylaxis Therapy 1 (PREEMPT 1) is a phase 3 study, with a 24-week, double-blind, parallel-group, placebo-controlled phase followed by a 32-week, open-label phase. Subjects were randomized (1:1) to injections every 12 weeks of onabotulinumtoxinA (155 U–195 U; n = 341) or placebo (n = 338) (two cycles). The primary endpoint was mean change from baseline in headache episode frequency at week 24.
Results: No significant between-group difference for onabotulinumtoxinA versus placebo was observed for the primary endpoint, headache episodes (−5.2 vs. −5.3; p = 0.344). Large within-group decreases from baseline were observed for all efficacy variables. Significant between-group differences for onabotulinumtoxinA were observed for the secondary endpoints, headache days (p = .006) and migraine days (p = 0.002). OnabotulinumtoxinA was safe and well tolerated, with few treatment-related adverse events. Few subjects discontinued due to adverse events.
Conclusions: There was no between-group difference for the primary endpoint, headache episodes. However, significant reductions from baseline were observed for onabotulinumtoxinA for headache and migraine days, cumulative hours of headache on headache days and frequency of moderate/severe headache days, which in turn reduced the burden of illness in adults with disabling chronic migraine.
Introduction
Chronic migraine (CM) is a complex, progressive, neurological disorder afflicting up to 2.4% of the general population (1–5). The condition is recognized as a complication of migraine in the the International Classification of Headache Disorders, second edition (ICHD-II) and is defined as experiencing headache on ≥15 days per month, of which ≥8 meet criteria for migraine without aura or respond to migraine-specific treatment (1,6). CM is a severely disabling condition and has been shown to significantly reduce patients’ health-related quality of life (HRQoL) (2,7).
Current treatment of CM may be complicated by the frequent use of acute headache pain medications, such as analgesics, triptans, opioids, or ergots (8–10). Controlled empirical data on the prophylactic treatment of chronic migraine is limited (11–14), and consequently there is little evidence-based medicine available to help physicians care for the patients (6). Currently there are no agents with regulatory approval as headache prophylaxis specifically for CM sufferers.
OnabotulinumtoxinA (BOTOX®; Allergan, Inc., Irvine, CA, USA) has been reported to relieve pain associated with a variety of conditions, including migraine headache (11,12,15–25). OnabotulinumtoxinA is thought to block peripheral signals to the central nervous system and indirectly inhibit central sensitization, leading to headache prophylaxis (26,27). Several placebo-controlled studies have evaluated the prophylactic potential of onabotulinumtoxinA in episodic migraine (migraine with <15 headache days per month) and chronic daily headache (≥15 headache days per month), including chronic tension-type headache (CTTH), with mixed efficacy results (11,12,20,28–32). The efficacy of onabotulinumtoxinA in episodic migraine and CTTH has not been established, although it is possible that patient selection criteria, and the dosage and injection paradigm used in these exploratory studies were not optimal (28–31). However, results from the chronic daily headache studies that predominantly included patients with CM suggested efficacy within this patient population and warranted further evaluation and confirmation (11,12).
The design of this comprehensive phase 3 program in CM took into consideration the previously identified issues (e.g., patient selection criteria, dosage, injection paradigm) from earlier exploratory studies (11,12,29,30), as well as the results of a promising post hoc analysis of an earlier phase 2 onabotulinumtoxinA chronic daily headache study (32). The PREEMPT (Phase III REsearch Evaluating Migraine Prophylaxis Therapy) clinical program (PREEMPT studies 1 and 2) was conducted to evaluate the safety and efficacy of onabotulinumtoxinA for headache prophylaxis in adults with CM. Results from the 24-week, double-blind, placebo-controlled phase of PREEMPT 1 are reported herein. PREEMPT 2 is reported in a companion manuscript.
Methods
Study design
The PREEMPT clinical program comprised two phase 3, multicenter, double-blind, placebo-controlled studies, PREEMPT 1 and PREEMPT 2. The PREEMPT 1 study was conducted from January 23, 2006, to July 16, 2008, at 56 North American sites. PREEMPT 1 was conducted to evaluate the efficacy and safety of onabotulinumtoxinA for prophylaxis of headaches in adults with CM. PREEMPT 1 had a 28-day baseline screening period (hereafter referred to as baseline) and a 24-week, double-blind, parallel-group, placebo-controlled phase with two injection cycles, followed by a 32-week, open-label phase with three injection cycles (Figure 1). An interactive voice response system (IVRS) daily telephone diary was used by patients to record their headache symptoms and acute treatments.
PREEMPT study design.
PREEMPT 1 was conducted in accordance with the Declaration of Helsinki ethical principles, Good Clinical Practices, principles of informed consent, and requirements of public registration of clinical trials in the United States (ClinicalTrials.gov identifier NCT00156910). The study was approved at each site by an independent ethics committee or a local institutional review board. Written informed consent was obtained from each randomized participant prior to any study-related procedures.
Study participants
Men or women aged 18 to 65 years with a history of migraine meeting the diagnostic criteria listed in ICHD-II (2004) section 1, migraine (1), with the exception of “complicated migraine” (i.e., hemiplegic migraine, basilar-type migraine, ophthalmoplegic migraine, migrainous infarction) were eligible. Randomized patients provided diary data on ≥20 of 28 days during baseline. They were required during baseline to have ≥15 headache days with each day consisting of ≥4 hours of continuous headache and with ≥50% of days being migraine or probable migraine days (referred to hereafter simply as migraine days); and ≥4 distinct headache episodes, each lasting ≥4 hours. Exclusion criteria included any medical condition that might put patients at increased risk if exposed to onabotulinumtoxinA (e.g., neuromuscular diseases); diagnosis of other primary or secondary headache disorders; use of any headache prophylactic medication within 28 days before start of baseline; a Beck Depression Inventory score of >24 at baseline; fibromyalgia; psychiatric disorders that could have interfered with study participation; or previous exposure to any botulinum neurotoxin serotype. Women of childbearing potential must have had a negative urine pregnancy test prior to each injection and have been using a reliable means of contraception.
Randomization, stratification and study treatment
Eligible patients were randomized in blinded fashion (1:1) to onabotulinumtoxinA treatment or placebo. Randomization was stratified based on the frequency of acute headache pain medication intake during the 28-day baseline as yes/no overuse of acute headache pain medications, where medication overuse–yes was defined as intake during baseline of simple analgesics on ≥15 days, or other medication types or combination of types for ≥10 days, with intake ≥2 days/week from the category of overuse. Treatments were balanced in blocks of four within each medication overuse stratum for each investigator site. Investigators were trained not to enroll patients who frequently used opioids as their acute headache pain medication. The randomization sequence was generated using SAS programming language (SAS Institute, Cary, NC, USA). Randomization programmers had access to the central server, where the randomization sequence was kept. The programmers then released the information to personnel who packed the medication kits, and to the vendor (Perceptive Informatics, Waltham, MA, USA) who managed the patient electronic diary for purposes of central implementation of the randomization and treatment-kit assignment. At the end of the baseline screening phase, when the investigator attempted randomization of a subject, the central implementation computer program determined if the subject met the quantitative inclusion/exclusion criteria as per the patient-reported diary data. If qualifications were met, the subject number was linked to the next randomization number grouped within strata for that site, and the site was notified of the medication kit assigned to that randomization number. Throughout the double-blind phase of the study, the patients, the investigators who administered the study treatment and assessed safety and outcomes, as well as the study sponsor management personnel were all masked to the treatment-group assignment.
The PREEMPT injection and dose paradigm was based on the experiences from phase 2 studies (11,12) and will be detailed elsewhere. OnabotulinumtoxinA 155 U or placebo was administered in 31 fixed-site, fixed-dose injections across seven specific head/neck muscle areas. At the investigator’s discretion, an additional 40 U could be administered into the temporalis, occipitalis and/or trapezius muscles using a follow-the-pain strategy. Decisions on additional dose and location were based on the patient’s report of usual location of predominant pain, severity of palpable muscle tenderness and clinician’s best judgment of the potential benefit of additional doses in the specified muscles. The maximum total dose was 195 U at 39 sites. Dosing and results in these studies are specific to the formulation of onabotulinumtoxinA manufactured by Allergan, Inc.
Efficacy and safety measures
The primary endpoint in PREEMPT 1 was mean change from baseline in frequency of headache episodes for the 28-day period ending with week 24. A headache episode was defined as patient-reported headache with a start and stop time indicating that the pain lasted ≥4 continuous hours. All other efficacy analyses were also based on the mean change from baseline to the 28 days ending with week 24. Prespecified secondary efficacy variables were frequency of headache days (defined as a calendar day [00:00 to 23:59] when the patient reported ≥4 continuous hours of headache diary episode), migraine days (defined as a calendar day with ≥4 continuous hours of headache meeting ICHD-II criteria for migraine 1.1, 1.2, or 1.6), migraine episodes (defined as patient-reported headache with a start and stop time indicating that the pain lasted ≥4 continuous hours and met ICHD-II criteria for migraine 1.1, 1.2, or 1.6), and overall acute headache pain medication use (all categories; referred to hereafter as acute pain medication intakes). Of note, in the PREEMPT 1 protocol and statistical analysis plan, the frequency of headache days was called out as the most important secondary efficacy measure because it was the only secondary efficacy measure that had prespecified sensitivity analyses, which included all those that were prespecified for the primary variable (headache episodes).
Other a priori defined analyses included three assessments of disability and HRQoL measured by the mean change from baseline at week 24 in total Headache Impact Test (HIT)-6 score (33), the Migraine Specific Quality of Life Questionnaire [MSQ v.2] (34,35) and the daily average of the Headache Impact score (HIS).
Statistical analysis
Planned enrollment for the study was approximately 650 patients. For headache episode frequency, the week 24 minimum retained sample size of n = 240 per group, with standard deviation of 5.5, would have >90% power to detect ≥1.75 between-group difference in mean change from baseline, using a two-sided alpha = 0.05. Due to the long duration of the study (56 weeks per patient), a larger sample size than what was needed for the week 24 primary efficacy analysis was planned to ensure sufficient sample size at the end of the study for long-term safety evaluations (>150 patients with five active treatment cycles).
All efficacy analyses used the intent-to-treat population, which included all randomized patients. For primary and secondary variables, the prespecified comparison between treatment groups was done by analysis of covariance of the change from baseline, with the same variable’s baseline value as covariate, with main effects of treatment group and medication overuse strata. The baseline covariate adjustment was prespecified as the primary analysis, but sensitivity analyses (e.g., rank-sum test on changes from baseline without a baseline covariate) were also performed. Scores for months with ≥20 days of diary data were prorated to 28-day equivalents. Scores for months with <10 days of diary data were estimated using a modified last observation carried forward (mLOCF) methodology. This involved the substitution of the patient’s previous 28-day period score multiplied by the ratio of the mean across all patients in the 28-day period of interest divided by the mean across all patients in the previous 28-day period. Scores for months with 10–19 days of diary data were estimated using an average of the prorated and mLOCF estimates. The mLOCF method of imputation of missing data was prespecified, but sensitivity analyses were also done (e.g., using observed data without imputation). For binomial variables, the between-group comparisons were done with Pearson’s Chi-square or Fisher’s exact tests, except that logistic regression with the same variable’s baseline as covariate was used for variables with baseline imbalance. A two-sided test with p ≤ .05 was considered to be statistically significant.
No control of the type-1 error rate for multiple secondary endpoints was prespecified in PREEMPT 1. Therefore, a highly conservative Bonferroni adjustment was applied to compare the week 24 p values to a critical level of .01, which adjusted the prespecified type-1 error rate of .05 for the five variables that were prespecified as primary or secondary.
Safety analysis was performed on all randomized patients who received at least one dose of study medication at day 0.
Results
Demographic and baseline headache characteristics
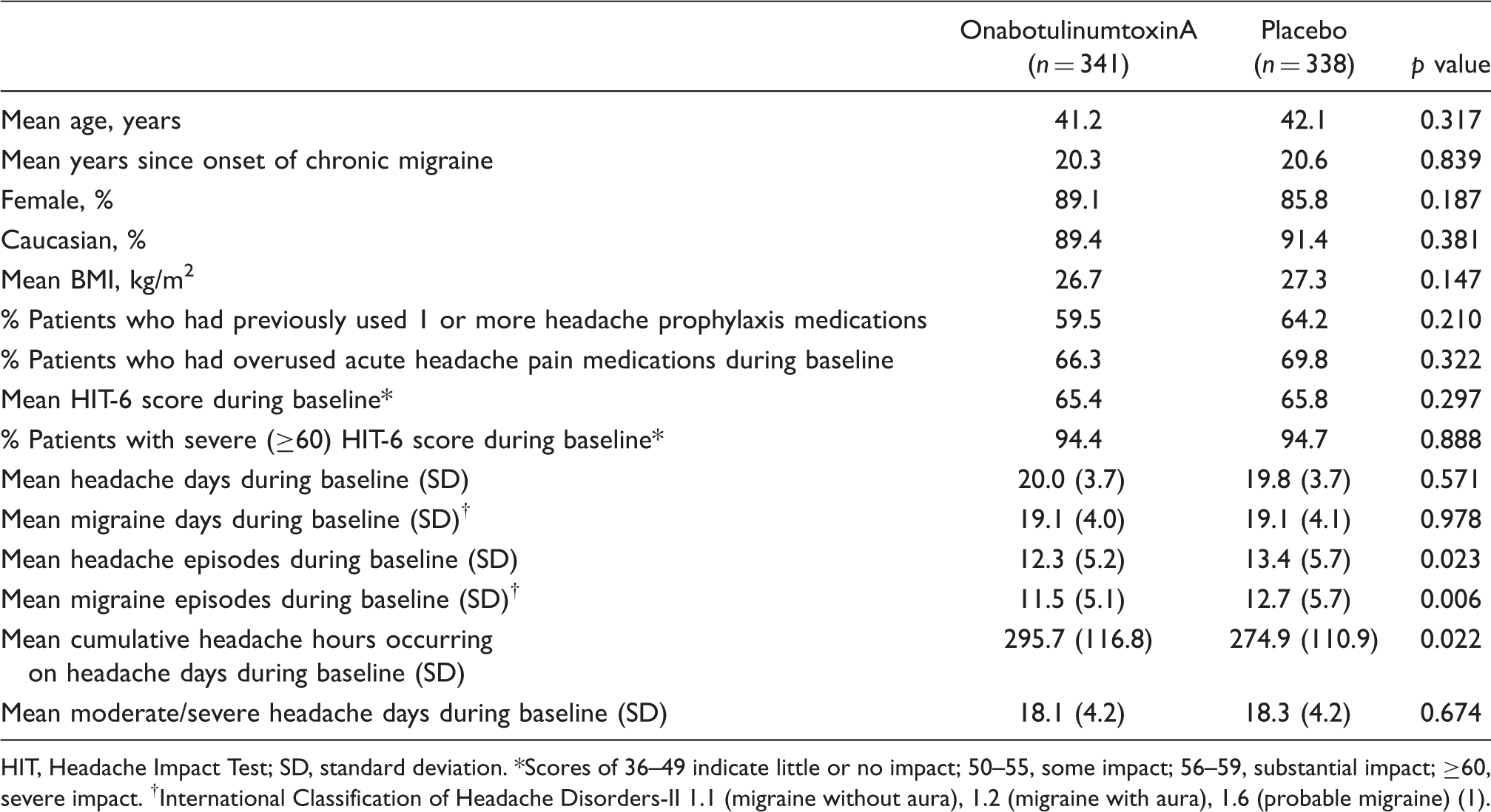
The recruitment period was between January 2006 and May 2007, with a 56-week follow-up period after the last patient was enrolled. Of 1713 patients screened, 679 were randomized to onabotulinumtoxinA (n = 341) or placebo (n = 338) (Figure 2). The majority of patients were female (87.5%) and Caucasian (90.4%); their mean age was 41.7 years. There were no significant between-group differences at baseline for the majority of important demographic characteristics (Table 1). However, at baseline, patients receiving onabotulinumtoxinA had significantly fewer headache episodes (12.3 onabotulinumtoxinA vs. 13.4 placebo; p = .023) and migraine episodes (11.5 onabotulinumtoxinA vs. 12.7 placebo; p = .006) than patients receiving placebo, and significantly more cumulative hours of headache occurring on headache days (p = .022). Most patients overused acute pain medications during the 28-day baseline. Few patients (71/679) used opioids during baseline; only 17 (2.5%) met opioid overuse criteria.
Patient disposition. Baseline demographics and characteristics HIT, Headache Impact Test; SD, standard deviation. *Scores of 36–49 indicate little or no impact; 50–55, some impact; 56–59, substantial impact; ≥60, severe impact. †International Classification of Headache Disorders-II 1.1 (migraine without aura), 1.2 (migraine with aura), 1.6 (probable migraine) (1).
Patient compliance rates in reporting data to the IVRS diary were high. Mean patient diary-day compliance rate at baseline was >99%, and the rate remained high (>93%) across both treatment groups over the 24-week, double-blind, placebo-controlled phase of the study.
Efficacy results
Primary endpoint: headache episodes. Despite a large within-group decrease from baseline, no between-group difference was observed for the primary endpoint, mean change from baseline in frequency of headache episodes at week 24 (−5.2 onabotulinumtoxinA vs. −5.3 placebo, p = .344; 95% confidence interval [CI] [−1.12, 0.39]) (Figure 3A; Table 2), based on the statistical model that used the prespecified baseline covariate adjustments. Prespecified sensitivity analyses (e.g., nonparametric) were also performed, but did not alter the outcome and produced similar results. We considered the possibility that the baseline imbalance of headache episode frequency between treatment groups, which is reported herein (.023) (Table 1), was an anomaly. An exploratory post hoc analysis of headache episode frequency showed no significant between-group difference during the first 14 days of baseline (p = .137) but did show a significant between-group difference during the last 14 days of baseline (p = .024). If the imbalance was an anomaly of randomly balanced groups, the change from baseline analysis could be misleading. Therefore, an analysis that used post-treatment counts (i.e., did not use baseline as a covariate and did not use changes from baseline) was performed. This post hoc analysis of headache episode frequency (not change from baseline) found significant between-group differences favouring onabotulinumtoxinA over placebo at week 4 (9.5, standard error [SE] ± 0.27; 10.5 SE ± 0.32; p = .015), week 8 (8.1, SE ± 0.28; 9.2 SE ± 0.32; p = .012), week 20 (7.3, SE ± 0.28; 8.3 SE ± 0.35; p = .05), and week 24 (7.3, SE ± 0.28; 8.1 SE ± 0.35; p = .049).
(A) Primary endpoint: mean change from baseline in frequency of headache episodes. Headache episode frequency at baseline: 12.3 ± 0.3 onabotulinumtoxin A group vs. 13.4 ± 0.3 placebo group; p = .023. All data presented as mean ± standard error. (B) Secondary variable: mean change from baseline in frequency of headache days. Headache day frequency at baseline: 20.0 ± 0.2 onabotulinumtoxinA group vs. 19.8 ± 0.2 placebo group, p = .571. All data presented as mean ± standard error. Efficacy of onabotulinumtoxinA at week 24 HIT, Headache Impact Test. *Primary efficacy endpoint. †International Classification of Headache Disorders-II 1.1 (migraine without aura), 1.2 (migraine with aura), 1.6 (probable migraine) (1). ‡Scores of 36–49 indicate little or no impact; 50–55, some impact; 56–59, substantial impact; ≥60, severe impact. 

Secondary efficacy endpoints. A large mean decrease from baseline with significant between-group difference for onabotulinumtoxinA was observed at all time points, including week 24, for the secondary efficacy variable frequency of headache days (−7.8 onabotulinumtoxinA vs. −6.4 placebo, p = .006; 95% CI [−2.40, −0.40]) (Figure 3B; Table 2) and migraine days (p = .002) (Table 2). A highly conservative Bonferroni adjustment for multiple comparisons at week 24 in which the critical level for p value comparisons was reduced from .05 to .01 did not alter the significance of these results (i.e., headache days and migraine days remained significantly improved versus placebo).
Large within-group improvements for mean change from baseline in frequencies of migraine episodes and acute pain medication intakes were observed, although neither showed significant between-group differences. A post hoc analysis conducted to identify potential patterns of intake by medication class (ergotamines, triptans, simple analgesics, opioids, combination analgesics, multiple analgesics) demonstrated that the frequency of triptan intake was significantly reduced from baseline in the onabotulinumtoxinA compared to the placebo group (−3.3 onabotulinumtoxinA vs. −2.5 placebo, p = .023) at week 24.
Post hoc analyses also found that onabotulinumtoxinA significantly reduced both cumulative hours of headache on headache days (p = .003) and frequency of moderate/severe headache days (p = .004) (Table 2).
Headache impact on functioning and HRQoL. OnabotulinumtoxinA-treated patients demonstrated a significant decrease in disability and improved functioning compared with placebo-treated patients as measured by mean change in total HIT-6 score at all time points (p < .001 at week 24; Table 2) and by proportion of patients with severe HIT-6 scores (p = .001). OnabotulinumtoxinA treatment statistically significantly improved HRQoL as measured by changes on the three MSQ role function domains: (i) restrictive (p < .001 at weeks 12 and 24); (ii) preventive (p = .005 at week 24); and (iii) emotional (p ≤ .002 at weeks 12 and 24). OnabotulinumtoxinA treatment also showed significant improvement versus placebo in HIS assessments at weeks 4, 8, 20 and 24 (p ≤ .029).
Safety and tolerability
Summary of overall adverse events reported in the 24-week double-blind phase

AE, adverse event. *Discontinuations during double-blind or open-label phases due to AEs with onset during the double-blind phase.
Discussion
Patients with CM present a clinical treatment challenge, and yet this population is almost always excluded from migraine prophylaxis trials. The PREEMPT patient cohort had on average suffered with frequent headache for two decades. Upon enrolling in this study, these patients experienced an average of 20 headache days per month and were highly disabled. These CM patients had been inadequately treated by available medical therapies, with approximately two-thirds having found any previous headache prophylactic medication treatments to be ineffective and/or intolerable. Two-thirds were overusing acute pain medication at baseline.
Despite the challenging clinical conditions posed by the PREEMPT cohort, treatment with onabotulinumtoxinA in PREEMPT 1 led to sustained improvements from baseline that favoured onabotulinumtoxinA treatment over placebo across multiple clinically relevant variables, including headache days, migraine days and several patient-reported quality of life measures. Though both groups improved from baseline, no between-group difference was noted for the primary endpoint, headache episodes. This is in contrast to significant findings for this endpoint for onabotulinumtoxinA reported by Diener et al. (36) in the second PREEMPT study. At baseline in this study (PREEMPT 1), the onabotulinumtoxinA group had both significantly fewer headache episodes (p = .023) and significantly longer cumulative headache duration (p = .022) than the placebo group (Table 1), which resulted in these patients having a mean of >20 cumulative headache hours more per month than those in the placebo arm. Post hoc analysis of PREEMPT 1 showed significant between-group differences favouring onabotulinumtoxinA treatment in frequency of headache episodes (the primary variable) at weeks 4, 8, 20, and 24 when treating the baseline imbalance as an anomaly. The variability in duration of headache episodes among migraine sufferers is well known, and this trial highlights the need for a more standardized endpoint such as headache days in future migraine trials.
In addition, in this study, onabotulinumtoxinA significantly reduced the frequency of headache days from baseline, which was the primary endpoint for PREEMPT 2 (36). Among secondary variables, significant differences favouring onabotulinumtoxinA over placebo were observed in frequency of migraine days, but not in frequency of acute pain medication intakes (all categories). There were both significant within-group decreases from baseline and a significant between-group difference favouring onabotulinumtoxinA for total cumulative hours of headache on headache days and moderate/severe headache days (Table 2). Importantly, this trial demonstrated that onabotulinumtoxinA may be effective in patients who overuse acute pain medication and who were considered treatment refractory based on past clinical experience. There were no significant differences favouring placebo for any efficacy variable at any time point in the study.
Although there were overall within-group reductions from baseline in acute pain medication intakes (all categories), there was no between-group difference. This apparent discrepancy between significant reductions in frequency of headache days but not in frequency of acute pain medication intakes may reflect patients’ use of medication for lower-grade headaches that did not qualify in level of intensity or duration (≥4 continuous hours) to be considered a headache day or episode according to the study eligibility and protocol definitions. Because acute pain medication (all categories) use was similar across onabotulinumtoxinA and placebo patient groups, significant improvements favouring onabotulinumtoxinA-treated patients across a number of efficacy measures evaluated during the study cannot be attributed to patterns of use of, or detoxification from, acute pain medication. Furthermore, investigators and coordinators in this trial were specifically instructed not to alter or counsel patients on the frequency or type of acute pain medication utilised during the course of the trial.
It is unlikely that the discrepancy between the observed frequency of headache days and the self-reported intake of acute pain medication use reflects inaccurate self-reporting, because patients only had to indicate yes or no concerning any day’s acute pain medication use, without providing additional details. An electronic diary system with a three-day maximum recall was selected for reporting in PREEMPT because it was known to be more reliable in this setting than a paper diary system. Patients were very compliant with diary self-reporting as evidenced by the >93% of patient self-reported daily diary data available for the week 24 primary time point evaluation.
Patients treated with onabotulinumtoxinA experienced significant improvements in their disability and their HRQoL. OnabotulinumtoxinA-treated patients reported improved functioning and vitality as well as reductions in psychological distress. The between-group comparison of mean change from baseline in HIT-6 disability score in favour of onabotulinumtoxinA treatment equaled the established clinically meaningful between-group minimum difference of 2.3 (37). Furthermore, improvement in the HRQoL of onabotulinumtoxinA-treated patients was observed in all three MSQ role function domains.
Treatment-related AEs were consistent with the known tolerability profile of onabotulinumtoxinA injected into head and neck muscles, and no newly emerging safety findings were observed. There were significantly more treatment-related AEs in the onabotulinumtoxinA group than in the placebo group. Overall, individual AEs occurred in fewer than 10% of patients, and were mild to moderate in severity and relatively short in duration.
Although there is no approved chronic migraine prophylaxis treatment that could have been included in PREEMPT as an active comparator (38), this gap is nonetheless a potential limitation in the PREEMPT program. The absence of such an active comparator precludes comparison of the efficacy of onabotulinumtoxinA with other headache prophylactic treatments. Comparisons of efficacy and safety of onabotulinumtoxinA treatment with other headache prophylactic treatments in the CM population will require head-to-head comparator trials. Also, a potential limitation of this study is the high placebo response rate. A large placebo response has been observed in migraine studies (39), which is consistent with our observations. In general, the placebo effect in pain trials such as this one can be attributed to expectation, Pavlovian conditioning, and reduction of anxiety (39). This phenomenon has been observed in prophylactic treatment studies involving episodic migraine, where higher variability is seen in the rate of the placebo response compared to acute migraine treatment (39). Parenteral pain treatments have higher placebo rates than placebo pills. Heightened expectations related to an injected treatment may be a factor influencing placebo response rates (28). One risk facing the trial was that the awareness of patients and/or investigators of physical changes that could occur (or were occurring) due to muscle relaxation in the forehead of patients treated with onabotulinumtoxinA would result in the potential unblinding of the study treatment. Although this phenomenon could have inflated the reporting of the active response, it is not consistent with a contrasting absence of a nocebo efficacy effect. In other words, patients could also have noticed an absence of physical change and been equally unblinded to placebo treatment, the basis for a nocebo response (lack of improvement from baseline when receiving placebo). Additionally, the presence of the placebo response suggests that the blind was maintained. Regression to the mean and/or spontaneous improvement are other possible explanations for the high placebo response.
The PREEMPT 1 study population is representative of the typical patient with CM seen in clinical practice, as demonstrated by population-based epidemiologic studies (40), and it can therefore be projected that physicians should expect similar results when treating patients with CM in the community. Results from PREEMPT 1 do not imply that onabotulinumtoxinA is effective in other patient populations that were specifically excluded from these trials, such as patients with episodic migraine, CTTH and secondary headache disorders.
PREEMPT 1 results demonstrated the superiority of onabotulinumtoxinA across multiple headache symptom efficacy measures, resulting in reduced disability and improved functioning, vitality, and overall HRQoL in adults with disabling CM. The PREEMPT 1 trial demonstrated that multiple treatments of 155–195 U of onabotulinumtoxinA per treatment cycle administered every 12 weeks is safe and well tolerated.
Disclosure
This study was sponsored by Allergan, Inc., Irvine, CA, USA.
Conflict of interest statement
SKA has received grants and research support from Advanced Bionics, Alexza, Allergan, Capnia, GlaxoSmithKline, MAP pharmaceuticals, Merck, Ortho-McNeil, Neuralieve, NuPathe and Takeda. She is a consultant for Ortho-McNeil, Merck, GlaxoSmithKline, Allergan, Neuralieve, NuPathe and MAP Pharmaceuticals. She has also received honoraria from Merck, GlaxoSmithKline, Kowa, NuPathe and Ortho-McNeil. DWD has received honoraria from Allergan, Merck, Neuralieve, Coherex, Kowa, Minster, NeurAxon, H Lundbeck, Endo, Pfizer, Nupathe and MAP Pharmaceuticals, in addition to being a consultant to and on the advisory board of these pharmaceutical companies. He has also received funding from Advanced Neurostimulation Systems, St. Jude Medical Center and Medtronic. CCT, RED and MFB are employees of Allergan, and own stock in the company. SDS and RBL have received honoraria and research funding from Allergan, in addition to being consultants to and on the advisory board of Allergan. HCD has received honoraria for participation in clinical trials, contribution to advisory boards, or oral presentations from Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, CoLucid, Bohringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Grunenthal, Janssen-Cilag, Lilly, La Roche, 3M Medica, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brummer, Sanofi-Aventis and Weber & Weber. He has also received financial support for research projects from Allergan, Almirall, AstraZeneca, Bayer, GlaxoSmithKline, Janssen-Cilag, and Pfizer. Headache research at the Department of Neurology in Essen, where HCD is Professor, is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF), and the European Union.
Footnotes
Acknowledgements
The authors would like to thank the patients who participated in the studies and their families. We also thank the Regional Lead Coordinating Investigators and Allergan Global Clinical Operations for their support, principal investigators and their staff for conducting the trials (PREEMPT 1 and PREEMPT 2) and current and past BOTOX® Headache Development advisory board members for their input and intellectual contributions to the development and advancement of the BOTOX® clinical development program. We would like to thank Allergan, Inc., for funding IntraMed Educational Group, New York, NY, to provide editorial support in the preparation and styling of this manuscript.
Werner J. Becker, Andrew Blumenfeld, F. Michael Cutrer, Astrid Gendolla, Christine Lay, Vince Martin, Maja Relja, Jack D. Schim, Timothy R. Smith, Thomas W. Ward, Paul Winner.
Jacqueline Abbas, Salim Ali, Billy Bahia, Steve Balyakin, Anne Blanco, Terry Boodhoo, Antoinette Brown, Roger Chan, Raymona Chin, George Demos, Christopher Dick, Dawn Drake, Ilana Fainaru, Laurie Fitch, Melissa Gilbert, Rozanno Gonzales, Amy Ho, Diane Hook, Courtney Huynh, Xiaofang Lei, Kimberlee Marruffo, Mary Ann Miller-Messana, David Nguyen, Dolly Nguyen, Shobhal Patel, Mimi Phomma, Stacey Quimpo, Anna Rask, Carminna Reyes, Mary Ann Robinson, Julie Roth, Regina Shin, Mai Sirimanne, Beatrix Taylor, Anisse Tse, Amanda VanDenburgh, Kathy Vu, Caroline Writer.
Sheena K. Aurora, Peter Barbour, Bing Behrens, Bradley Boop, Jan Brandes, Michael Brown, Walter Carlini, Suzanne Christie, Paula Crenshaw, Fred Cutrer, Khashayar Dashtipour, Robert Duarte, Richard Dubinsky, Aaron Ellenboger, Stephen Forner, Marshall Freeman, Frederick Freitag, Rose Giammarco, Matthews Gwynn, Jessica Heiring, Neal Hermanowicz, Allan Herskowitz, Steven Herzog, Steven Hong, Marc Husid, Hubert Leonard, Kenneth Levin, Morris Levin, Ivan Lopez, Robert Loss, Lisa Mannix, Herbert Markley, Ninan Mathew, Alexander Mauskop, Peter McAllister, Martin Mollen, May Ong-Lam, Patrick Parcells, James Patton, Eric Pearlman, R. Allan Purdy, Dennis Riff, Lawrence Robbins, Michael Rossen, John Rothrock, Jack Schim, Harvey Schwartz, Todd Schwedt, Stephen Silberstein, Richard Singer, Stuart Stark, Barbara Vogler, Christopher Voll, Robert Vollbracht, Jeanette Wendt, Anthony Wheeler, Kerri Wilks, Paul Winner, Bradley Wrubel, Hemant Yagnick.
Sheena K. Aurora, Werner J. Becker, Andrew Blumenfeld, Rami Burstein, Carl Dahlof, Hans-Christoph Diener, David M. Dodick, Anthony W. Fox, Fred G. Freitag, Richard B. Lipton, Ninan T. Mathew, Jane Osterhaus, Julio Pascual, Joel R. Saper, Stephen D. Silberstein, K.M.A. Welch, Paul Winner.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.