
Abstract
The kidney plays an important role in iron homeostasis and mesangial cells (MCs) are phagocytic cells important for glomerular homeostasis. Sickle hemoglobin (HbS) modulators are promising clinical candidates for treatment of sickle cell disease. Although they prevent disease pathophysiology of HbS polymerization and red blood cell (RBC) sickling by increasing hemoglobin oxygen affinity, higher oxygen affinity can also cause transient tissue hypoxia with compensatory increases in erythropoiesis and subsequent increases in RBC turnover. CD-1 mice treated with an HbS modulator for 2 weeks developed higher RBC mass, increased erythropoiesis, and, by 1 month, deposition of intracellular pigments in renal tubular and parietal epithelium. In addition, in mice treated for 26 weeks, pigment was observed in MCs, which was accompanied by glomerular cell aggregates (MC hypercellularity) and tubulo-interstitial inflammation. The pigment was confirmed by Perl’s iron staining and transmission electron microscopy (TEM) to be iron-containing proteins. Glomerular cell aggregates were confirmed to be MCs by TEM, and Ki-67 immunolabeling suggested that MC hypercellularity was due to proliferation. Collectively, these findings, along with iron-containing proteins in livers and spleens, suggested that iron overload secondary to increased RBC turnover led to increased renal iron reabsorption. While both MC hypercellularity and tubulo-interstitial inflammation were thought to be responses to long-term accumulation of iron, the former was considered a homeostatic response to eliminate iron, and maintain glomerular structure and function, while the latter was more consistent with an iron-catalyzed oxidative stress response. To our knowledge, this is the first report of MC hypercellularity in a preclinical toxicity study.
Keywords
Sickle cell disease is the most common inherited hematologic disorder and affects over 7 million people worldwide. 9 The disease is caused by a missense mutation in the β-globin gene of hemoglobin (Hb) giving rise to sickled hemoglobin (HbS). HbS polymerizes more readily in the deoxygenated state leading to distorted sickle-shaped RBCs that occlude capillaries (vaso-occlusion) and prevent tissue oxygen delivery (hypoxia-reperfusion injury). These effects lead to episodes of pain, hemolytic anemia, stroke, and related pathologies. 23 Since oxygenated HbS does not polymerize, identification of compounds that bind to HbS and allosterically modulate the oxygenation state has been an area of therapeutic focus for Sickle cell disease. Considerable efforts have been made to develop HbS modulators that can stabilize oxy-HbS. However, only a few of these HbS modulators are currently being used clinically.10,23
The test article used in the studies described in this article is a noncovalent HbS modulator that binds to Hb with nanomolar affinity. 10 In a 2-week multiple dose study using Towens sickle cell disease mice, the test article caused significant stabilization of the oxy-Hb state. Stabilization of the oxy-Hb state in the test article treated mice translated into 37.8% reductions in RBC sickling as well as improvement in markers of hemolytic anemia compared with vehicle treated mice.10,15 The test article achieved consistently high levels of Hb occupancy (> 40%) in animals, which was sustained for the duration of dosing. The chemical structure and details about absorption, distribution, metabolism, and excretion of the compound are discussed elsewhere.10,15
Although of therapeutic benefit for sickle cell disease, one of the consequences of increased Hb-oxygen affinity is lower release of oxygen leading to transient tissue hypoxia. 16 This is especially true with the high doses used in toxicity studies. Across different species, hypoxic conditions can stimulate erythropoiesis by stabilizing hypoxia inducible factors that, in turn, upregulate erythropoiesis-stimulating hormone erythropoietin (EPO) production by the kidney.8,14,32 Hypoxia inducible factors also enable iron utilization for increased erythropoiesis by upregulating the expression of many iron-transport proteins including divalent metal transporter-1, ferroportin (FPN), and transferrin receptor-1. EPO and erythroferrone, a hormone produced by erythroblasts, also inhibits hepcidin synthesis in the liver, which enables iron utilization for increased erythropoiesis under hypoxic conditions in humans and mice.13,14
About 70% of the iron in mammals is found in the oxygen transport protein Hb in RBCs. Hence, iron is critical for RBC synthesis, including erythropoiesis in medullary (bone marrow) and extramedullary (spleen or liver) tissues. On the contrary, iron present in the Hb of senescent RBCs is efficiently reused and recycled during erythrophagocytosis by macrophages of the reticuloendothelial system in the liver and spleen.1,28,32 Under physiological conditions, most circulating iron [Ferric or Fe (III)] is bound to serum protein transferrin. In cases of systemic iron overload, when the iron binding capacity of transferrin is exceeded, iron binds loosely to other carriers, including albumin and citrate as nontransferrin-bound iron. Since iron is bound loosely to carriers as nontransferrin-bound iron, it is readily available as a cellular labile (or catalytic) iron pool composed of redox-active iron (Fe2+), which can participate in redox cycling (the Fenton reaction). 32 Fenton reactions release reactive oxygen species (ROS) that can cause oxidative stress (oxidative damage to cell, membranes, proteins, and DNA), ferroptosis (cell death associated with iron-dependent oxidative stress), and inflammation.21,32 At lower physiological intracellular levels, however, ROS has beneficial effects and activates redox signaling or initiates cell proliferation and differentiation (oxidative eustress). 26 Hence, several organs and cellular compartments communicate in a coordinated way to regulate the concentration of iron in biological fluid to provide iron as needed and avoid iron toxicity (iron homeostasis).
The kidney plays a key role in systemic iron hemostasis across species by preventing iron loss from the body by means of reabsorption.8,31,32 Both transferrin-bound iron and nontransferrin-bound iron are filtered by the glomerulus into the renal tubular lumen. Subsequently, the bulk of filtered transferrin-bound iron is reabsorbed by endocytosis at the apical surfaces of proximal and distal tubule epithelial cells via receptors, such as transferrin receptor-1 or megalin-cubilin complex. Ferric or divalent iron [Fe(II)] is reabsorbed by divalent metal transporter-1 and zinc transporter ZIP8 and/or ZIP14, and Hb is reabsorbed by the megalin-cubilin complex in both humans and mouse models. 32 Accordingly, systemic iron deficiency and eventually anemia can be observed in cases of renal disorders caused by reduced iron reabsorption/increased urinary iron loss and increased glomerular leakage of iron. 32
Once inside the cell, iron can be exported to the systemic circulation, utilized, or safely stored. Cytosolic iron is transported back to systemic circulation via exporter FPN located at the basolateral surface of proximal tubule epithelial cells.8,32 Hepcidin, a peptide hormone produced by the liver and involved in systemic iron homeostasis, can block iron export by inducing FPN internalization and degradation. 8 Intracellular iron can be utilized by the mitochondrion to synthesize several iron-containing proteins, including heme, aconitase, and cytochromes. Nonutilized and nonexported iron inside the cell can be stored safely as Fe(III) in ferritin complexes or in hemosiderin (denatured ferritin with amorphous protein and lipid formed usually under iron overload).12,17
A nephron, the functional unit of the mammalian kidney, consists of a blood-filtering unit, the glomerulus, and a reabsorption unit, the tubules. The MCs together with podocytes and glomerular endothelial cells form the glomerular capillary tuft, which is responsible for the kidney’s ultrafiltration function. Parietal epithelial cells that form the outer layer of Bowman’s capsule are the fourth cell type within the glomerulus.3,22 MCs make up about 30% to 40% of the glomerular cell population and produce a mesangial matrix. The mesangium (MCs together with matrix) forms a stalk that holds and organizes the multiple capillary loops within the glomerulus.2,3 Accordingly, injury or loss of MCs, as occurs with mesangiolysis, leads to loss or simplification of capillary loops. MCs are considered glomerular stromal cells and in addition to supporting glomerular capillaries and maintaining homeostatic functions, they have phagocytic activity. As such, they are involved in glomerular development and the production of glomerular basement membrane matrix, and may facilitate glomerular basement membrane turnover.2,6 Once activated, MCs can regulate immune responses (recruitment of immune cells through secretion of cytokines/chemokines or expression of Toll-like/intracellular pattern recognition receptors), control inflammation, and mediate the reparative response to injury. 2
Aberrant expansion of mesangium (MC hypercellularity and matrix accumulation) has been observed in certain renal diseases as a response to abnormal amounts of debris accumulating in the glomerular filtration membrane, such as diabetic nephropathy, immunoglobulin (Ig) A nephropathy, focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis, and lupus nephritis in humans and animal models.18,29,33 –35 In these diseases, however, expanded mesangium is associated with other glomerular lesions (eg, focal necrosis, segmental scarring, crescents in Bowman’s space, glomerular basement membrane thickening, or immune cell infiltration) and/or tubulo-interstitial and vascular changes (interstitial fibrosis, tubular atrophy, interstitial inflammation, vascular sclerosis, or tubular red cell casts/ proteinaceous casts).7,18,35 MC hypercellularity is an uncommon lesion and with time and further injury, increased production of mesangial matrix proteins and proteinuria can lead to various glomerular and tubular lesions.7,18 In humans, glomerular lesions including MC hyperplasia have been observed in several malignant diseases although primary tumors of glomeruli are rare and not well documented.
Here, we characterize renal lesions including MC hypercellularity, pigment accumulation in glomerular and tubular cells, and tubulo-interstitial inflammation as well as associated hematological parameters in toxicity studies conducted in CD-1 mice with an HbS modulator. The relationship between these renal lesions and likely pathogenesis are discussed.
Materials and Methods
Animals and Husbandry
CD-1 mice (Charles River Laboratories Stone Ridge, New York) that were 6-week-old at the start of dosing were randomly assigned to 4 groups. There were 3 studies conducted in mice, of 2 weeks, 1 month, and 26 weeks of dosing duration, respectively. For each study, 3 groups of mice/sex were given an HbS modulator once daily by oral gavage at 3 different doses (low, mid, and high) for 2 weeks (study 1; 10 mice/sex/group), 4 weeks followed by 1-month recovery (study 2; 10 mice/sex/group for dosing phase and 5 mice/sex/group for recovery phase), and 26 weeks followed by a 3-month recovery (study 3; 15 mice/sex/group for dosing phase and 10 mice/sex in mid-dose group for recovery phase). One separate group of an equal number of mice was given 2% (w/v) polyvinylpyrrolidone K30 (PVP K30) and 1% (v/v) polysorbate 80 (Tween 80) and served as controls for dosing and recovery phases of each study.
Mice were housed individually in polycarbonate cages in environmentally controlled rooms (68–79°F, relative humidity 30–70%, 12-hour light/12-hour dark photoperiod). Mice were provided Certified Irradiated Rodent Diet 5002 (PMI Feeds Inc.) and water ad libitum.
Toxicology studies were conducted in facilities accredited by the American Association for Accreditation of Laboratory Animal Care, and in accordance with regulations and established guidelines reviewed and approved by an Institutional Animal Care and Use Committee.
Pathology
Clinical laboratory parameters were evaluated in samples collected from all toxicity animals. Blood smears from all animals were prepared and blood cell morphology was evaluated microscopically. For anatomic pathology parameters, mice were euthanized with CO2 following an overnight fast and were necropsied at the end of the dosing phase (all 3 studies) and recovery phase (studies 2 and 3). Organ weights from a standard panel of organs were collected. All major organs were fixed by immersion in 10% neutral-buffered formalin, with the exception of the eyes, which were collected and fixed in 3% glutaraldehyde and then transferred to neutral-buffered 10% formalin. Tissues were routinely processed to paraffin blocks, and hematoxylin and eosin-stained slides were prepared. The following additional histochemical stains were performed using conventional methods to characterize pigment in the kidney: Perl’s iron stain for iron, Hall’s for bile pigments, and Schmorl’s for lipofuscin. A subset of livers and spleens were also sectioned and stained for Perl’s iron. All slides were permanently mounted with coverslips and then scanned into a digital database using the Aperio AT2 (Leica Biosystems, Buffalo Grove, Illinois). Microscopic lesions were graded on a scale of 1 to 5 as minimal, mild, moderate, marked, or severe, based on the extent of change detected in the microscopic sections. Clinical pathology parameters and anatomic pathology (slides) were independently evaluated by 3 board certified pathologists.
TEM
Formalin-fixed mouse kidneys were trimmed to 2 mm cubes and post-fixed in Karnovsky’s fixative (2.5% glutaraldehyde/2% paraformaldehyde in 0.1M cacodylate buffer pH 7.4). Tissue samples were washed 3× for 10 minutes with 0.1 M buffer and then post-fixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 2 hours at 25°C. Following osmication, tissues were washed 2× with deionized water, dehydrated through a graded alcohol series, transitioned through pure acetone, and then infiltrated in a 50:50 mixture of acetone and Poly/Bed 812 Embedding Media (Hard formula) overnight at room temperature. Dehydration and infiltration were carried out on a shaking platform. Samples were changed into pure resin 3× and infiltrated for at least 2 hours each, prior to embedding. Samples were embedded into silicone molds and cured for at least 24 hours at 60°C.
Resin blocks were sectioned on a Leica UC7 ultramicrotome at 0.5 µm, mounted on glass slides, and stained with 1% toluidine blue. After evaluation, blocks containing regions of interest were selected, thin sectioned at 75 nm, and mounted on copper grids. Thin sections were evaluated using a Hitachi HT-7820 transmission electron microscope (TEM) and images were captured using a BioSprint digital CCD camera system (Advanced Microscopy Techniques, Danvers, MA).
Ki-67 Immunohistochemistry and Image Analysis
Formalin-fixed, paraffin-embedded tissue sections were mounted on Super Frost Plus slides (12-550-15, Thermo Fisher Scientific, Waltham, MA) at a thickness of 5 µm for immunohistochemistry (IHC) analysis. IHC was performed on the Leica Bond RX (Leica Biosystems) using a rabbit monoclonal antibody to Ki67 (clone: SP6, catalog number: 275-16, Cell Marque/Sigma, Rocklin, CA). Sections were deparaffinized and pretreated with Epitope Retrieval 2 (AR9640, Leica Biosystems) for 20 minutes at 100°C. Slides were incubated in peroxide block using the Bond DAB Polymer Refine Detection Kit (DS9800, Leica Biosystems) for 5 minutes followed by primary antibody incubation at 0.53 µg/ml for 30 minutes. Ki-67 was detected using the anti-rabbit horseradish peroxidase (HRP) polymer for 8 minutes and DAB chromogen for 10 minutes in the Bond DAB Polymer Refine Detection kit. A positive tissue control for Ki-67 was included in the IHC run, as well as a rabbit IgG isotype control antibody at a matching concentration (I-1000, Vector Laboratories, Newark, CA). All slides were counterstained with hematoxylin, dehydrated through graded ethanol and xylenes, and permanently mounted with coverslips.
A deep learning analysis protocol package was trained in Visiopharm (Version 2023.09x64) with a DeepLabsV3 framework to identify glomeruli in Ki-67 IHC sections and label them as regions of interest. 4 Kidney sections from 3 animals per group were analyzed with 894 glomeruli were identified and classified in the control group and 1042 in the high-dose group. Within the glomerular regions of interest, a second linear Bayesian classifier analysis protocol package was applied to identify and quantify cells that were positive for Ki-67 expression as well as glomerular tufts that contained positive cells. Graph plotting and statistical analyses were performed in Graphpad Prism software (version 10.2.1). A t test was performed to test for significant difference between the groups.
Results
There was no test article-related mortality during the dosing or recovery phase of 3 toxicity studies.
Two-Week Study
There were no test article-related organ weight changes, gross pathology findings, or microscopic findings in the kidney. Test article-related microscopic findings were observed in the bone marrow (increased erythroid cellularity) and the spleen (extramedullary hematopoiesis) at the mid and/or high dose (Supplemental Table S1). Increased cellularity of erythroid cells in the bone marrow was characterized by prominent and more frequent clusters of erythroid precursors. Extramedullary hematopoiesis in the spleen was characterized by increased clusters to sheets of hematopoietic cells within the red pulp; the white pulp was not affected.
Hematology findings included increased RBC mass (Hb, hematocrit, and RBCs), mean cell volume (MCV), mean cell Hb (MCH), reticulocytes, and RBC distribution width (RDW) in mid- and/or high-dose animals (Supplemental Table S2). Blood smear examination revealed higher incidence and severity of polychromasia and anisocytosis among high-dose animals, which correlated with reticulocytosis. Hematological findings correlated with microscopic findings in the bone marrow and spleen.
One-Month Study
There were no test article-related gross pathology findings or organ weight changes in the kidneys at the time of necropsy. Microscopic findings in the kidneys were limited to minimal to mild golden-brown granular intracytoplasmic pigments predominantly in proximal tubular epithelium and, to a lesser degree, in parietal epithelium of the glomerulus in both sexes at the mid and high doses (Supplemental Table S3). In addition to the higher incidence and/or severity of bone marrow and spleen findings, mid- and/or high-dose mice in the 1-month study had extramedullary hematopoiesis in the liver (Supplemental Table S3). Incidence and severity of pigment deposition in the renal tubular epithelium at the high dose reduced at the end of 1-month recovery phase suggesting a partial recovery of this finding. All other microscopic findings completely recovered at the end of the 1-month recovery phase.
Hematological findings were similar to the 2-week study (Supplemental Table S4). Except for increased RDW, all hematological findings completely recovered at the end of a 1-month recovery phase.
Twenty-Six-Week Study
There were no test article-related gross pathology findings or organ weight changes in the kidneys at the time of necropsy. An incidence summary of relevant microscopic findings is presented in Table 1.
Summary of relevant dosing phase microscopic findings in CD-1 mice from a 26-week toxicity study.
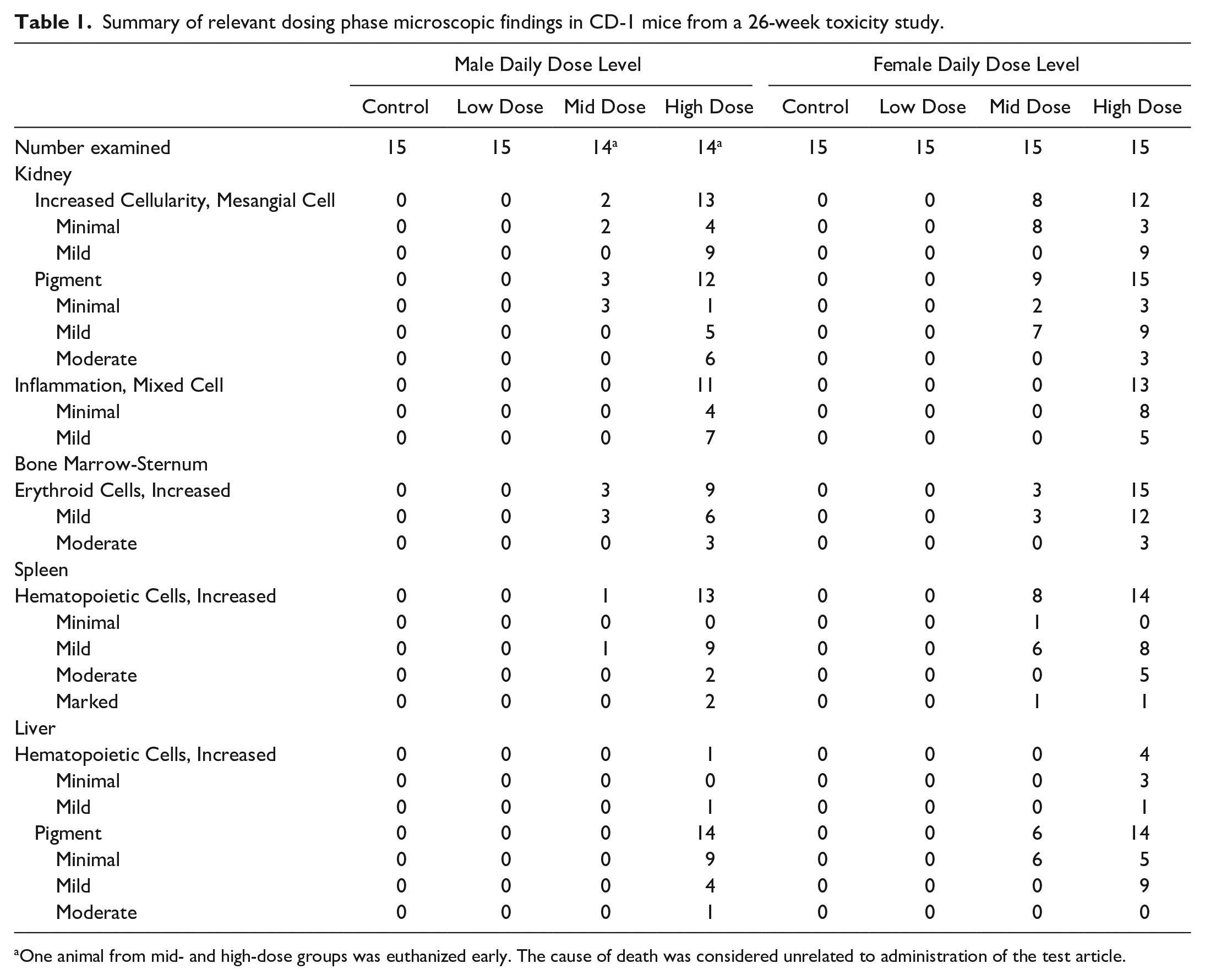
One animal from mid- and high-dose groups was euthanized early. The cause of death was considered unrelated to administration of the test article.
Increased cellularity of MCs characterized by aggregation of cells within the glomerular tufts was observed in the mid- and high-dose mice. MCs appeared as one to multiple, round to elongated, globular basophilic structures under light microscopy (Fig. 1a-c). TEM evaluation confirmed increased numbers of MCs embedded in the expanded mesangial matrix; cells frequently contained bilobulated or multilobulated nuclei (Fig. 2). Other components of the glomerular tuft, including cells (podocytes, endothelial cells), endothelial fenestrations, and podocyte foot processes, appeared intact and no infiltrating mononuclear or polymorphonuclear leukocytes were observed. In addition, Ki-67 immunolabeling demonstrated significantly higher numbers of positive nuclei in the glomerular tufts of high-dose mice compared with very few Ki-67 positive nuclei in controls. Notably, the Ki-67 positive glomerular cells in the high-dose animals were often located within MC aggregates (Fig. 3a, b).
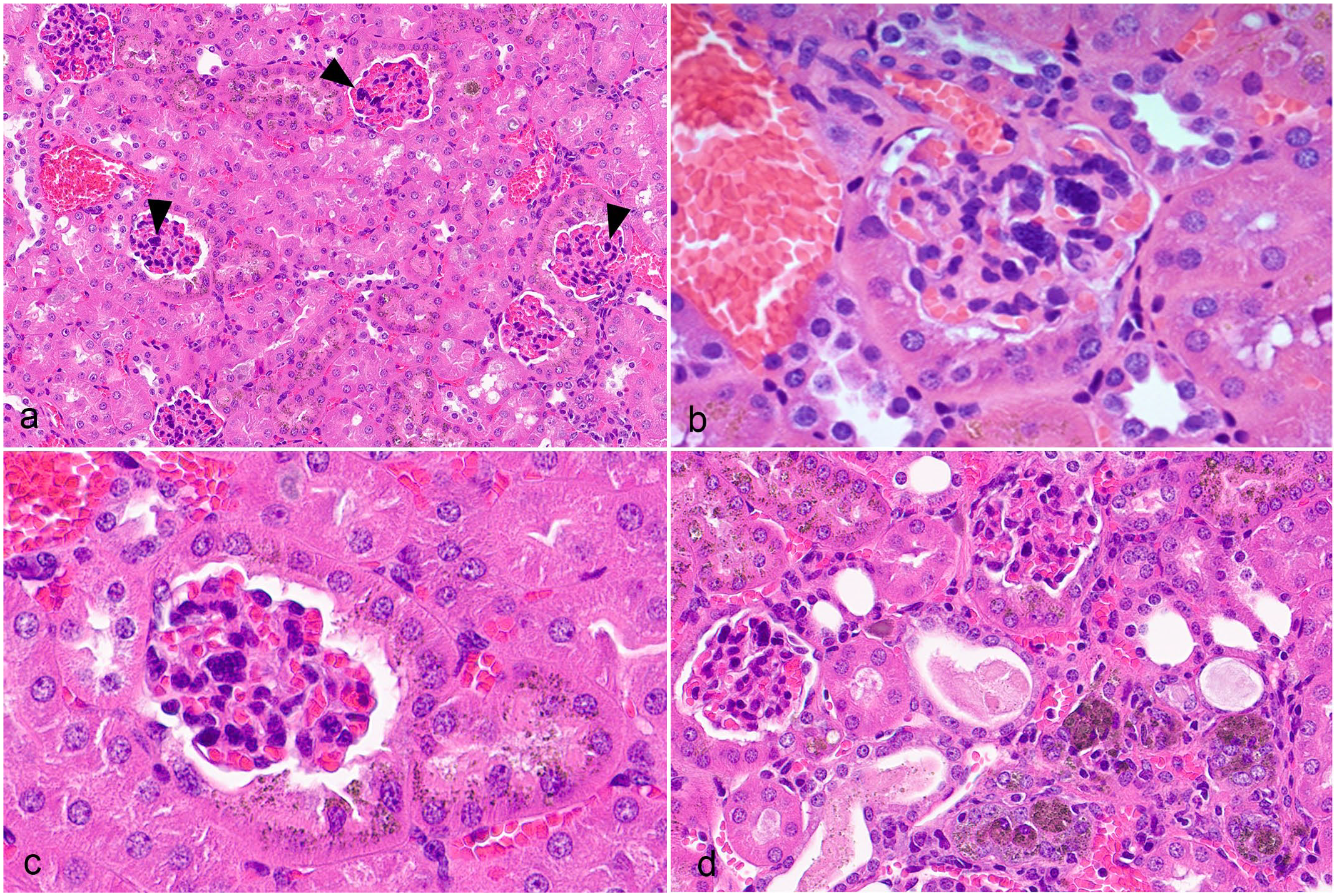
Kidneys from CD-1 mice following 26-week treatment with a sickle hemoglobin modulator showing glomerular and tubular lesions. Hematoxylin and eosin (HE). (a) Cell aggregates in the glomerular tufts (arrowheads). (b, c) Higher magnification showing aggregates of cells within the glomerular tuft (b, c) and pigment accumulation in the parietal and adjacent tubular epithelial cells (c). (d) Inflammatory cells infiltrate in the tubular and interstitial areas. A few tubular lumens are filled with debris, and golden-brown pigments can be seen within tubule epithelial cells and macrophages.
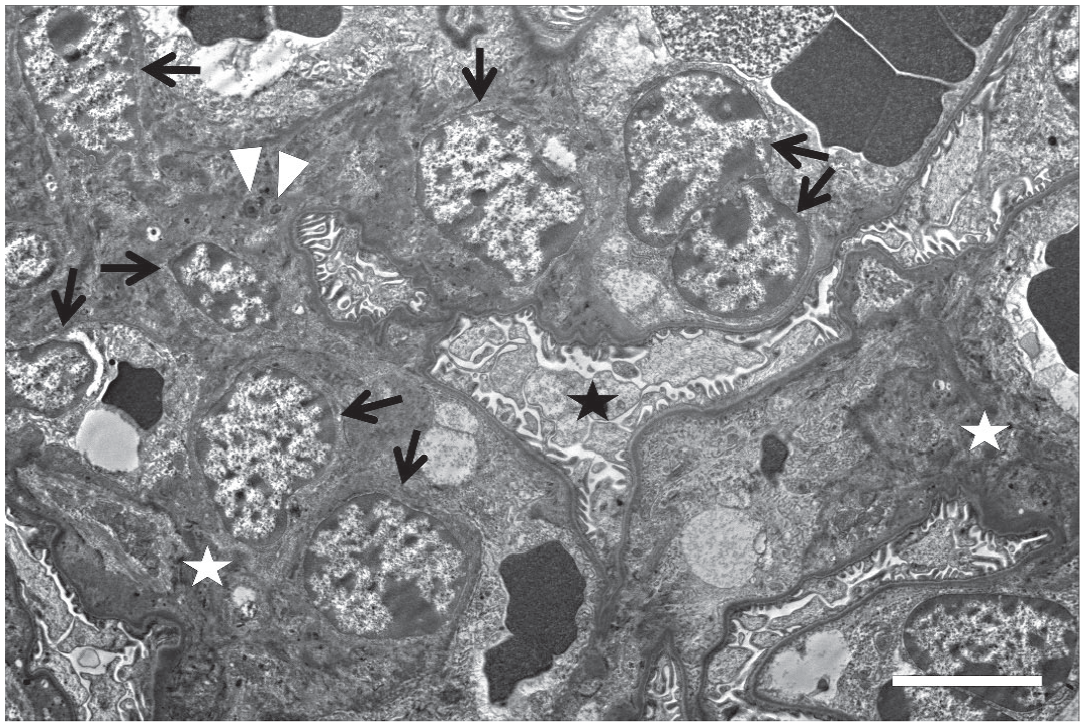
Transmission electron microscopy (TEM) of mesangial cell (MC) hypercellularity in the kidney of CD-1 mice following 26-week treatment with a sickle hemoglobin modulator. TEM image of a glomerulus showing proliferating MCs (black arrows) with occasional bi-nucleated cells. Prominent siderosomes containing electron dense fine granules can be seen within the cytoplasm of MCs (white arrowheads). Bar = 5 µm. White asterisks, mesangial matrix; black asterisk, foot processes.
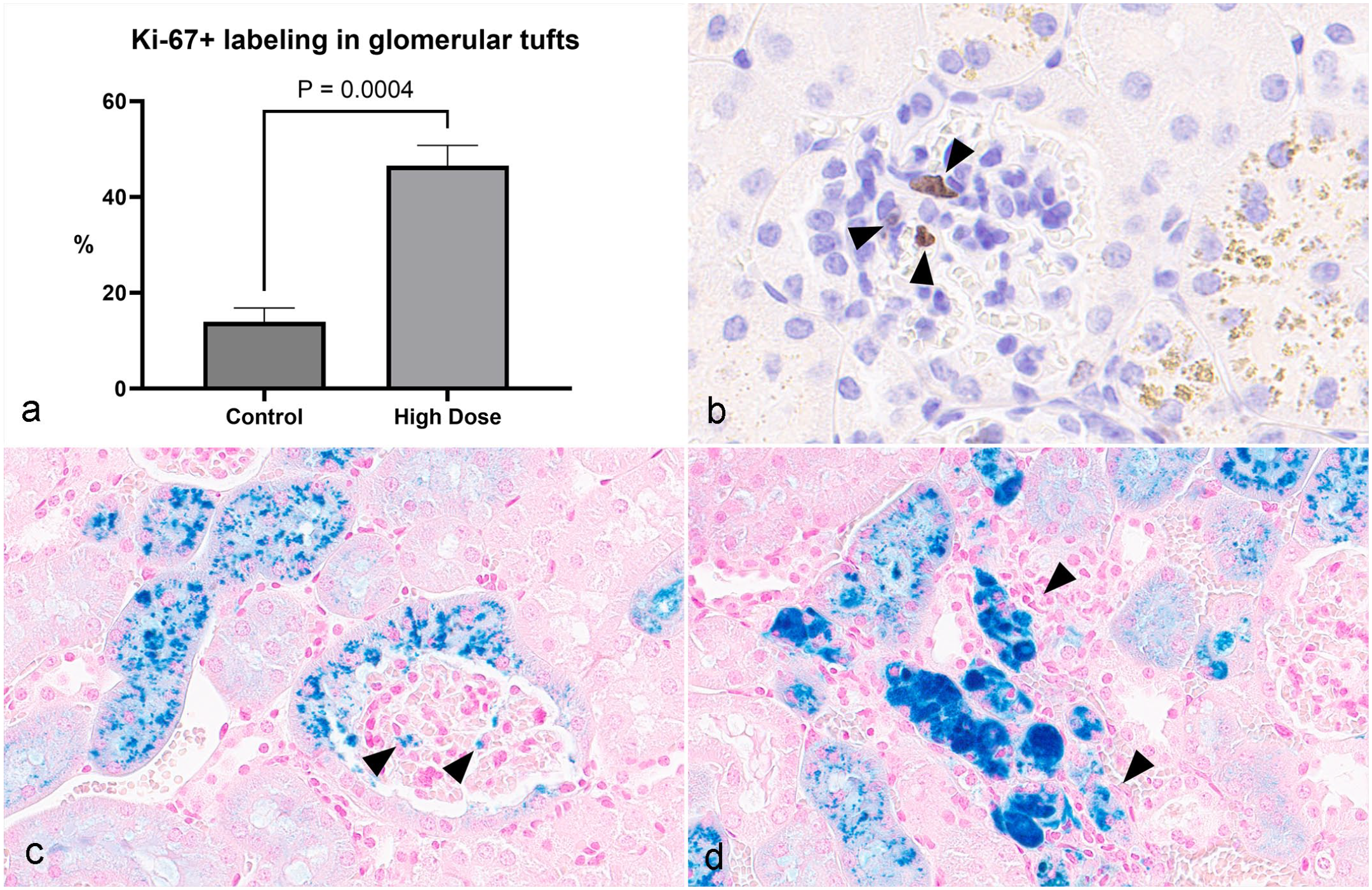
Ki-67 immunohistochemistry and Perl’s iron staining of renal cells from CD-1 mice following 26-week treatment with a sickle hemoglobin modulator. (a) High-dose mice exhibited significantly more Ki-67 immunolabeling in glomerular tuft cells compared with controls (n = 3). b) Cells within aggregates of mesangial cells in the glomerular tuft have Ki-67 immunolabeling (arrowheads). (c, d) Golden-brown pigments in the parietal cells and tubule epithelial cells are positive for Perl’s iron staining. Scattered cells in the glomerular tufts (c) and intact/degenerate tubule epithelial cells and inflammatory cells in the areas of inflammation (d) are stained positive with Perl’s iron (arrowheads).
Similar to the 1-month study, granular golden-brown pigments were observed in the glomerular parietal epithelial cells (Fig. 1c) and the adjacent epithelium of proximal and distal convoluted tubules (Fig. 1c, d). These pigments stained positive with Perl’s iron stain, indicative of iron pigments (Fig. 3c, d), and negative with Schmorl’s stain for lipofuscin and Hall’s stain for bile (data not shown). TEM evaluation revealed electron dense aggregates scattered within the cytoplasm or within siderosomes (consistent with iron accumulation) of glomerular parietal epithelial cells, MCs, and tubular epithelial cells (Fig. 4a, d).
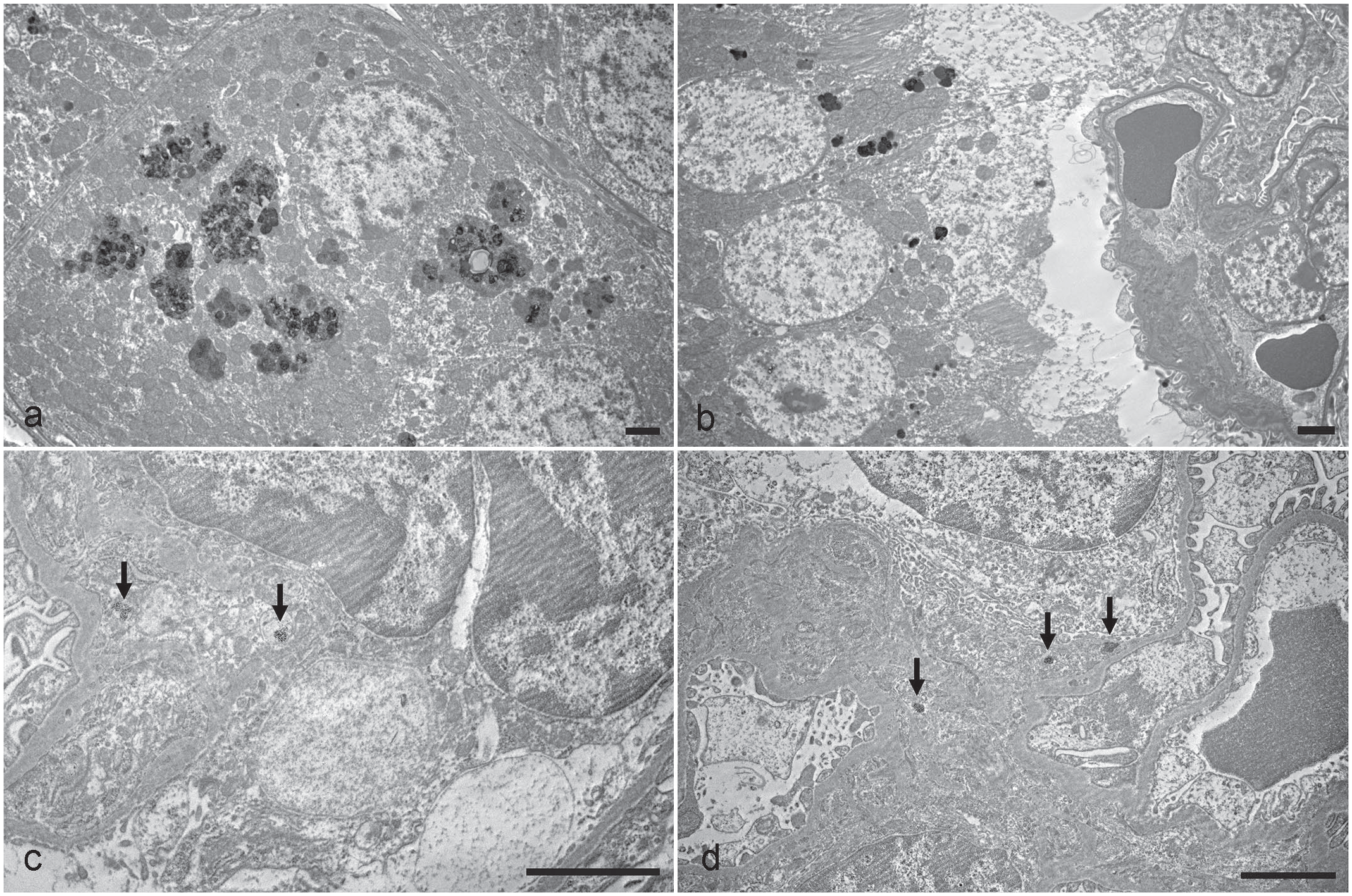
Transmission electron microscopy of renal cells from CD-1 mice following 26-week treatment with a sickle hemoglobin modulator. (a, b) Prominent siderosomes containing electron dense fine granules in the cytoplasm of a tubule epithelial cells (a) and parietal cell (b). (c, d) siderosomes (arrows) in the cytoplasm of mesangial cells (c) and in the mesangial matrix (d). Bars = 2 µm.
High-dose males and females also had multifocal mixed cell tubulo-interstitial inflammation characterized by small numbers of lymphocytes, plasma cells, and macrophages and fewer neutrophils (Fig. 1d). The tubules adjacent to the areas of inflammation were often basophilic and were occasionally lined with flattened degenerative epithelial cells; some tubular lumens contained mixed and degenerative inflammatory cells. Frequently, macrophages and tubule epithelial cells within the areas of inflammation contained golden-brown pigments (Fig. 1d).
In addition to the higher incidence and/or severity of the bone marrow, spleen, and liver microscopic findings, mid and/or high-dose mice in 26-week study had pigment accumulation in Kupffer cells and sinusoidal endothelial cells in the liver and pigment accumulation in the sinusoidal macrophages in the spleen. The pigment in the hepatic Kupffer cells and endothelial cells and splenic macrophages was confirmed by Perl’s iron staining to be iron-containing proteins (Fig. 5a, b).
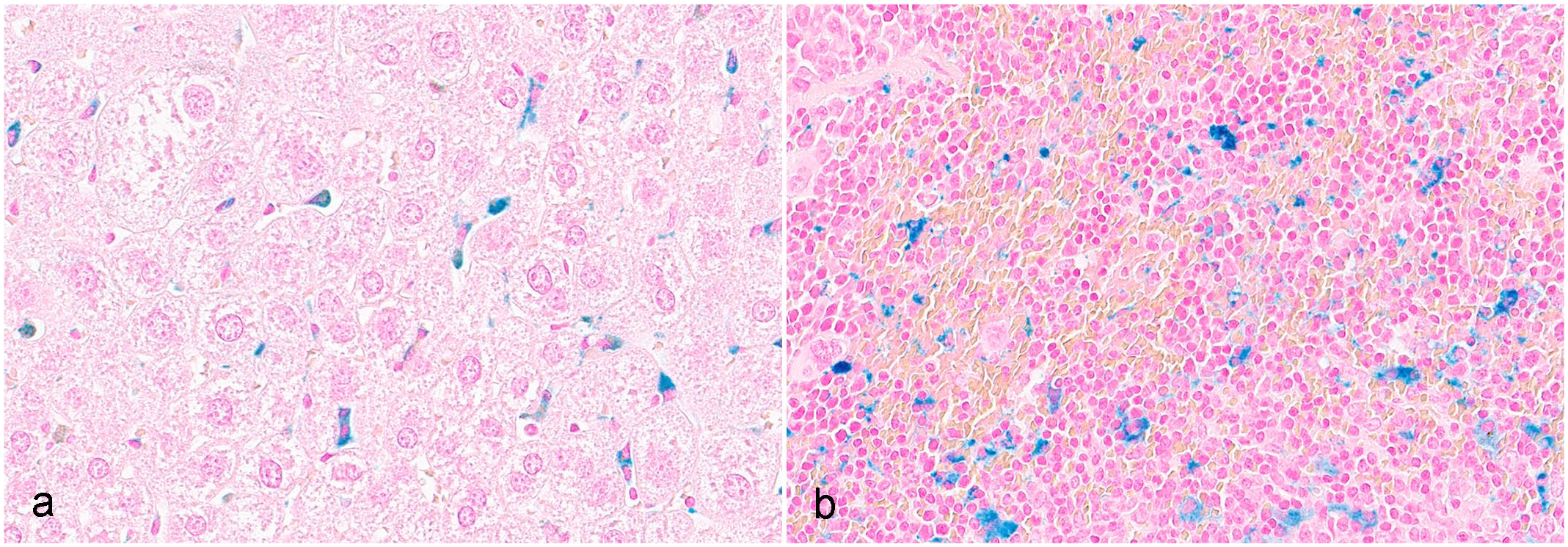
Perl’s iron staining of the liver and spleen of CD-1 mice following 26-week treatment with a sickle hemoglobin modulator. (a) Kupffer cells in the liver and (b) sinusoidal macrophages in the spleen are positive for iron with Perl’s stain.
Hematological findings were similar to the 2-week and 1-month studies (Table 2). Clinical pathology observations that could indicate an effect on the kidneys (eg, blood urea nitrogen or creatinine, urinalysis findings) were not present. All microscopic findings and hematological changes were considered nonadverse based on small magnitude of change, absence of adverse clinical correlates, and/or lack of associated microscopic or clinical pathology findings.
Summary of relevant dosing phase hematology parameter effects (mean control values and ratio relative to control) in CD-1 mice from a 26-week toxicity study.
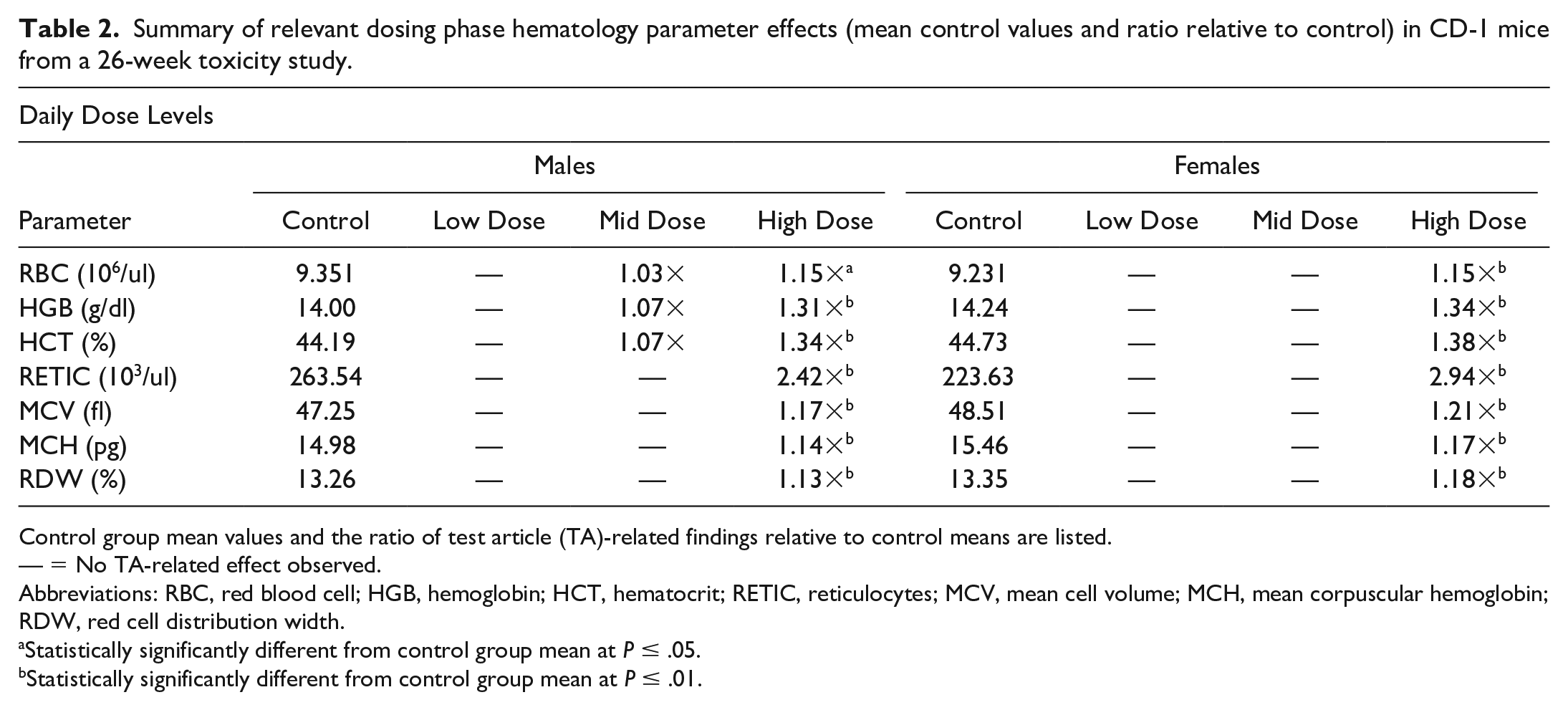
Control group mean values and the ratio of test article (TA)-related findings relative to control means are listed.
— = No TA-related effect observed.
Abbreviations: RBC, red blood cell; HGB, hemoglobin; HCT, hematocrit; RETIC, reticulocytes; MCV, mean cell volume; MCH, mean corpuscular hemoglobin; RDW, red cell distribution width.
Statistically significantly different from control group mean at P ≤ .05.
Statistically significantly different from control group mean at P ≤ .01.
The reversibility of the dosing phase findings was assessed after a 3-month recovery period (vehicle control and mid-dose groups only). All dosing phase hematological changes (higher RBC count, Hb concentration, and hematocrit) and all dosing phase microscopic findings at the mid-dose in the liver, spleen, bone marrow, and kidney (inflammation) completely reversed, except for the increased cellularity of MCs and pigments in the kidney, which exhibited partial recovery. Reversibility was only assessed at the mid dose (and not the low and high doses) given that anticipated exposure multiples at this dose were deemed sufficient to support further clinical development of the compound.
Discussion
An HbS modulator was evaluated in 2-week, 1-month, and 26-week toxicology studies in CD-1 mice. While MC hypercellularity and inflammation were observed only in the 26-week study, the incidence and severity of intracytoplasmic pigment accumulation in the kidney was higher in the 26-week study than the 1-month study. Pigment accumulation in the liver and spleen was also observed only in the 26-week study. Hematological findings (higher red cell mass parameters and higher reticulocytes), on the contrary, were observed in all 3 studies and correlated with increased erythropoiesis in the hematopoietic system (bone marrow, spleen, and liver).
These pathology findings indicate that the changes initially occurred in the hematopoietic system (increased erythropoiesis) and then subsequently involved circulating blood cells (higher red cell mass parameters and higher reticulocytes), the liver and spleen (increased RBC destruction in the reticuloendothelial system), and the kidneys (iron accumulation leading to inflammation and MC hypercellularity) (Fig. 6). This sequence of events aligns well with the pharmacology of the test article, which is known to increase oxygen affinity of Hb leading to tissue hypoxia.10,15 In this context, the observed findings were generally consistent with compensatory physiologic responses initiated to increase oxygen delivery to tissues. Increased EPO production by stabilized hypoxia inducible factors under hypoxic conditions can lead to increased erythropoiesis.8,32
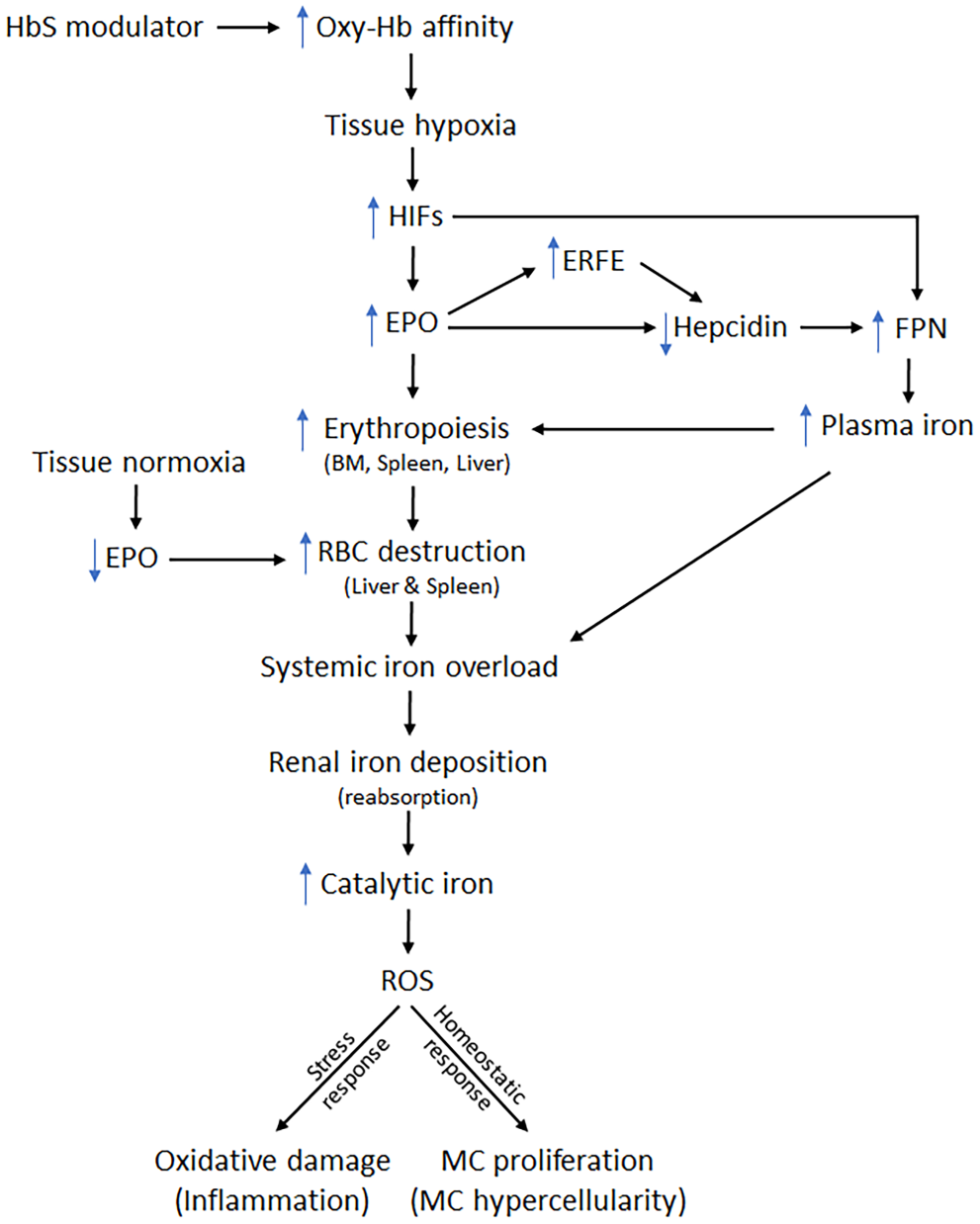
Flow chart showing hypothesized series of events leading to the tubulo-interstitial inflammation and mesangial cell proliferation upon sickle hemoglobin (HbS) modulator treatment for 26 weeks in mice. HIFs, hypoxia inducible factors; ERFE, erythroferrone; EPO, erythropoietin, FPN, ferroportin; BM, bone marrow; RBC, red blood cell; ROS, reactive oxygen species; MC, mesangial cell.
Increased RBC turnover related to pharmacologically induced increases in erythropoiesis was associated with increased RBC destruction. The resulting increase in iron turnover can then result in excess free iron, which subsequently accumulates as golden-brown pigment in organs known to be involved with iron metabolism (kidney, liver, and spleen). These pigments were interpreted to be iron-containing proteins (ferritin and/or hemosiderin) based on positive staining with Perl’s iron and the TEM findings of electron dense aggregates in the cytosol or within siderosomes.12,17 Accumulation of iron or iron-containing proteins primarily in the Kupffer cells of the liver and sinusoidal macrophages of the spleen further indicated RBC phagocytosis and increased breakdown.12,28 Stimulated erythropoiesis has been shown to be associated with decreased average RBC age. 20 In addition, young RBCs have increased metabolic activity and a lower Hb-O2-affinity than senescent RBCs, and they also have higher membrane fluidity and deformability, which eases the passage of blood through capillaries. 19 Recent studies have revealed a role for EPO in both RBC production and RBC destruction. EPO released during transient hypoxia would increase the production of new RBCs whereas a drop in the concentration of EPO at the return to normoxia would reduce the lifespan of circulating RBCs. 1 Accordingly, a higher RBC turnover characterized by both increased destruction of RBCs and increased production of young RBCs (indicated by increased reticulocytes) can be observed during increased erythropoiesis in healthy individuals. Given the longer life span of RBCs in mice (~40 days), 30 we did not observe any iron accumulation suggestive of RBC breakdown in the 2-week study. However, progressive iron accumulation in the kidney in longer-term studies (1 month and 26 weeks) suggested increased RBC turnover leading to iron overload and reabsorption from the glomerular filtrate. Increased labile iron associated with iron overload would further reduce RBC lifespan due to oxidative stress (RBC membrane damage by oxygen radicals and subsequent phagocytosis). Apart from increased RBC turnover, excessive iron absorption in the blood secondary to inhibition of hepcidin mediated FPN destruction by hypoxia-induced EPO or erythroferrone might also have contributed to iron overload.8,32
In the kidney, accumulation of iron deposits was mostly observed in tubule epithelial cells (primarily in the proximal tubule cells) and also in glomerular cells to a lesser extent. Iron accumulation in tubular epithelial cells was consistent with their reabsorption and storage function in iron hemostasis (reabsorption, storage, and recycle). Tubule epithelial cells, especially proximal tubular epithelial cells have all necessary iron-handling proteins (receptors, exporters, proteins association with metabolism or storage); these cells can reabsorb free and bound forms of iron as well as heme/Hb. 32 Among glomerular cells, iron deposits were frequently observed by light microscopy and TEM in the parietal cells lining Bowman’s capsule. To our knowledge, iron accumulation in glomerular parietal cells has not been reported and no iron-handling proteins specific to glomerular parietal cells have been identified. Given that these flat parietal cells abruptly transition to cuboidal epithelial cells of the proximal convoluted tubule at the urinary pole, and that these 2 cell types share a common mesenchymal origin, 22 it is possible that iron-handling proteins in the parietal cells are similar to tubule epithelial cells, which enables them to reabsorb iron from glomerular filtrate in the urinary space.
In addition to pigment accumulation, 2 additional renal findings in the 26-week CD-1 mouse study included tubulo-interstitial inflammation and increased cellularity of MCs. Tubulo-interstitial inflammation in high-dose males and females was likely caused by iron overload and associated elevated concentration of labile or catalytic iron in tubule epithelial cells and/or interstitial macrophages. Multiple studies suggest long-term exposure of the kidney to increased iron levels can result in kidney injury by oxidative damage known as ferroptosis (acute kidney injury or chronic kidney disease).21,32 Intracellular catalytic iron has been shown to stimulate inflammatory responses in macrophages whereas advanced oxidation proteins produced by the ROS (Fenton reaction) in kidney cells can cause inflammation by activating proinflammatory cytokines (eg, IL-6). 32 ROS can also reduce proximal tubule epithelial cell viability and proliferation in humans and animals leading to oxidative cellular injury. 31 In fact, we saw iron deposits within tubule epithelial cells and macrophages in areas of inflammation and cell death by both hematoxylin and eosin staining and Perl’s iron staining, indicating a causal relationship between inflammation and iron.
Although the exact mechanism for MC hypercellularity is unclear, elevated labile/catalytic iron associated with iron overload in glomerular filtrate was likely responsible for this proliferative renal change. ROS produced by labile iron in the Fenton reaction can induce lipid oxidation, 32 and oxidized lipoproteins have been shown to promote MC proliferation via activating the EGFR/MAPK pathway and preventing apoptosis. 5 Indeed, the significantly higher Ki-67 immunolabeling of cells (often in cellular aggregates) in the glomerular tuft of test article treated mice compared with controls in the 26-week study indicated that the MC hypercellularity was due to an increase in MC proliferation. 25 Higher MC proliferation resulted in MC hypercellularity. This was likely an oxidative eustress response unlike the oxidative stress/damage response observed in the tubulo-interstitium. In addition to maintaining glomerular structure and glomerular filtration rate,2,27,29,36 MCs have phagocytic activity, and they might function in vivo to clean up the glomerular filter of pathogens and deposited extra-glomerular material.2,6 Several pathways involved in phagocytosis have been identified in both mouse and human MCs, and these cells showed high phagocytic activity ex vivo.2,11 We observed scattered iron deposits in the cytoplasm and within siderosomes of the proliferating MCs by TEM. Unlike the inflammatory changes in the tubule interstitium, there were no mononuclear or polymorphonuclear leukocyte infiltrates in the glomeruli or evidence of structural damage to glomerular tuft components or cells. Furthermore, MC hypercellularity partially recovered at the end of a recovery period. Thus, it is plausible to assume that glomerular MC hypercellularity observed in the 26-week chronic mouse study was not a direct effect of the test article but a homeostatic response to maintain glomerular structure and function by eliminating accumulated iron or iron-containing proteins including Hb in the glomerular filtrate. Although it was not clear if the observed MC iron deposits were secondary to receptor mediated endocytosis or active phagocytosis, it is known that MCs have iron receptors (eg, transferrin receptor-1) as well as iron regulatory proteins (IRP1 and heme oxygenase 1) that may allow these cells to reabsorb iron from glomerular filtrate and store them in ferritin complexes. 32
Toxicity of this HbS modular was also evaluated in beagle dogs (24 to 36-month-old at the start of study) in 3 studies (same duration and similar doses) (data not shown). Similar to CD-1 mice, findings were mainly observed in the kidney (iron pigmentation), liver (iron pigmentation), hematopoietic system (erythropoiesis), and blood (increased RBC parameters). However, despite the overlapping systemic concentrations of the HbS modulator, MC hypercellularity and tubulo-interstitial inflammation were not observed in any of the dog studies including one that was 26-weeks in duration. Although, the exact mechanism for the species difference in renal findings is not known, this might be related to the longer RBC life span in dogs (~115 days in dogs vs ~40 days in mice) and the lower level of random RBC removal in dogs compared with mice.24,30 These species differences would result in comparatively lower RBC destruction and labile iron generation in dogs. Consistent with this assumption, we observed reduced severity and/or incidence of pigment accumulation in the kidney of dogs relative to mice in both the 1-month and 26-week studies. In addition, a species difference in the effect of the test article on Hb-oxygen affinity (eg, lower affinity in dogs) may have contributed to differences in microscopic pathology in the kidney.
In summary, in vivo studies in CD-1 mice demonstrated that administration of an HbS modulator induced changes in the hematopoietic system including hematology parameters (higher red cell mass parameters and higher reticulocytes) with subsequent effects in the kidney. These changes were not considered adverse, and all findings demonstrated either complete or partial recovery. These changes were in the kidney were considered secondary effects related to pharmacological effects of the test article on the hematopoietic system. In addition, our data indicated that mice are more sensitive than dogs in the development of proliferative MC changes.
Supplemental Material
sj-pdf-1-vet-10.1177_03009858241306400 – Supplemental material for Mesangial cell hypercellularity and iron accumulation in the kidney associated with administration of a sickle hemoglobin modulator in CD-1 mice
Supplemental material, sj-pdf-1-vet-10.1177_03009858241306400 for Mesangial cell hypercellularity and iron accumulation in the kidney associated with administration of a sickle hemoglobin modulator in CD-1 mice by Shambhunath Choudhary, Catherine Picut, Sarah R. Vargas, Diana Otis, Timothy M. Coskran, David Karanian, Jamie K. DaSilva, Christopher Houle and Laurence O Whiteley in Veterinary Pathology
Footnotes
Acknowledgements
The authors thank the staff at the Charles River Laboratories (CRL) Inc., Spencerville and Pfizer Inc., Groton, for their assistance in necropsy and sample collection.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are current or former employees of Pfizer Inc. and CRL Inc. and may own Pfizer’s or CRL’s stock.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Pfizer is the sponsor of the study.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.