
Abstract
The development of mouse models that replicate the genetic and pathological features of human disease is important in preclinical research because these types of models enable the completion of meaningful pharmacokinetic, safety, and efficacy studies. Numerous relevant mouse models of human disease have been discovered in high-throughput screening programs, but there are important specific phenotypes revealed by histopathology that are not reliably detected by any other physiological or behavioral screening tests. As part of comprehensive phenotypic analyses of over 4000 knockout (KO) mice, histopathology identified 12 lines of KO mice with lesions indicative of an autosomal recessive myopathy. This report includes a brief summary of histological and other findings in these 12 lines. Notably, the inverted screen test detected muscle weakness in only 4 of these 12 lines (Scyl1, Plpp7, Chkb, and Asnsd1), all 4 of which have been previously recognized and published. In contrast, 6 of 8 KO lines showing negative or inconclusive findings on the inverted screen test (Plppr2, Pnpla7, Tenm1, Srpk3, Sidt2, Yif1b, Mrs2, and Pnpla2) had not been previously identified as having myopathies. These findings support the need to include histopathology in phenotype screening protocols in order to identify novel genetic myopathies that are not clinically evident or not detected by the inverted screen test.
Discovering the genetic origins of disease is crucial to understanding the underlying pathogenetic mechanisms and in developing mutation-specific therapies. The development of mouse models that replicate the genetic and pathological features of human disease is vital in preclinical research because these types of models enable the completion of meaningful pharmacokinetic, safety, and efficacy studies.
From 2000 through 2008, Lexicon Pharmaceuticals performed high-throughput mouse knockout (KO) and comprehensive phenotypic analyses (Genome5000) for >4650 genes 7 in order to identify pharmaceutically relevant targets for treating human disease. 61,63,64 In the course of phenotyping these genetically engineered mice, a great deal of fundamental information on gene function and genetic diseases was first defined in mice and later confirmed in humans. 8 The KO strategies employed (described below) involved both gene trapping 15 using the OmniBank I embryonic stem (ES) cell library, and homologous recombination technologies. Phenotyping assays included a battery of tests in the areas of behavior, cardiology, immunology, metabolism, oncology, and ophthalmology, and included serum chemistry, and a high-fat diet obesity challenge. Lexicon’s high-throughput screening protocol included histopathologic evaluation of viable KO lines.
Numerous relevant mouse models of human disease have been discovered in screening programs performed by laboratories participating in the International Mouse Phenotyping Consortium (IMPC). 29 However, although histopathology is clearly required to fully characterize mutant mice in high-throughput phenotypic screening programs, 2 it is not routinely completed by many of the laboratories participating in the IMPC. Fortunately, IMPC laboratories are collecting and storing a standardized biobank of paraffin-embedded tissues for each mouse line 2 but the identification of many potentially important disease models is being delayed until histopathology is completed.
In Lexicon Pharmaceuticals’ phenotyping program, histopathology identified numerous phenotypes that were not detected by any other physiological or behavioral screening tests. In fact, histopathology actually proved to be the most efficient and sensitive method for detecting phenotypes in the upper respiratory tract, teeth, eyes, muscle, and kidney. 44 For example, although some tooth abnormalities may be visible grossly, KO phenotypes characterized by significant enamel or dentin defects were only detected histologically. 52,54 Detection of tooth phenotypes by histopathology is rapid and cost-effective because the cross-sections of the nose that are included in standard histopathology tissue sets include incisor and molar teeth in addition to the nasal passageways. At the same time, evaluation of the same nasal sections permitted rapid diagnosis of motile ciliopathies in mice, which usually present with clinically asymptomatic rhinosinusitis. 51,53,56 In the eye, retinal degeneration can be detected at very early stages by histopathology, 45,50 which is especially valuable in high-throughput phenotyping programs that generally evaluate relatively young mice in order to reduce husbandry costs. In addition, histopathology of the eye is probably the most sensitive approach for detecting pigmentation defects in mice. 55 In this report, we present evidence that histopathology is also the most sensitive and specific assay available for identifying otherwise-undetectable myopathies in genetically engineered mice. Histopathology also provides information that can help elucidate the underlying pathomechanisms most likely to be involved in the development of these myopathies.
Materials and Methods
Phenotype Screening
Knockout and littermate wild-type (WT) control mice were subjected to a comprehensive battery of phenotype screening assays as previously described. 7,8,61,63 In addition to histopathology, these screening assays included behavioral tests (such as circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, urinalysis, quantitative magnetic resonance, dual-energy X-ray absorptiometry scans, computed axial tomography (CAT) scans, microcomputed tomography (micro-CT) scans, fertility testing, and skin fibroblast proliferation assays. Immunology assays included hematology, peripheral blood flow cytometric analysis, acute phase response, and ovalbumin challenge.
Mouse Production
For all KO lines included in this report, the complete allele symbols are provided in Table 1. However, for brevity, only the basic gene symbols are used in the text. Lexicon utilized both gene trapping and homologous recombination methods to generate KO mice for the Genome 5000 project. Gene trapped lines were derived from strain 129S5SvEvBrd–derived ES cells obtained from the OmniBank I library, 1 as described previously. 61 To achieve effective gene disruption when using gene-trap mutations, intragenic insertions intersecting all known transcript units were selected after identifying the precise location of vector insertion using inverse genomic polymerase chain reaction (PCR). Oligonucleotide primers complementary to the gene-trap vector were used to amplify the vector insertion site for each clone, which was then compared to mouse genome sequence assemblies to localize the insertion with respect to the exons and introns of the gene. Gene disruption in vivo for gene-trap mutations was confirmed by a direct analysis of gene expression using reverse transcriptase (RT)-PCR. RNA was extracted from at least 2 tissues of WT and homozygous mutant mice using a bead homogenizer and RNAzol (Ambion) according to manufacturer’s instructions. Reverse transcription was performed with SuperScript II (Invitrogen) and random hexamer primers, according to the manufacturer’s instructions. PCR amplification was performed with oligonucleotide primers complementary to exons flanking the insertion site.
Genes and antemortem phenotype of 12 knockout mouse lines with histopathologically detected myopathies.
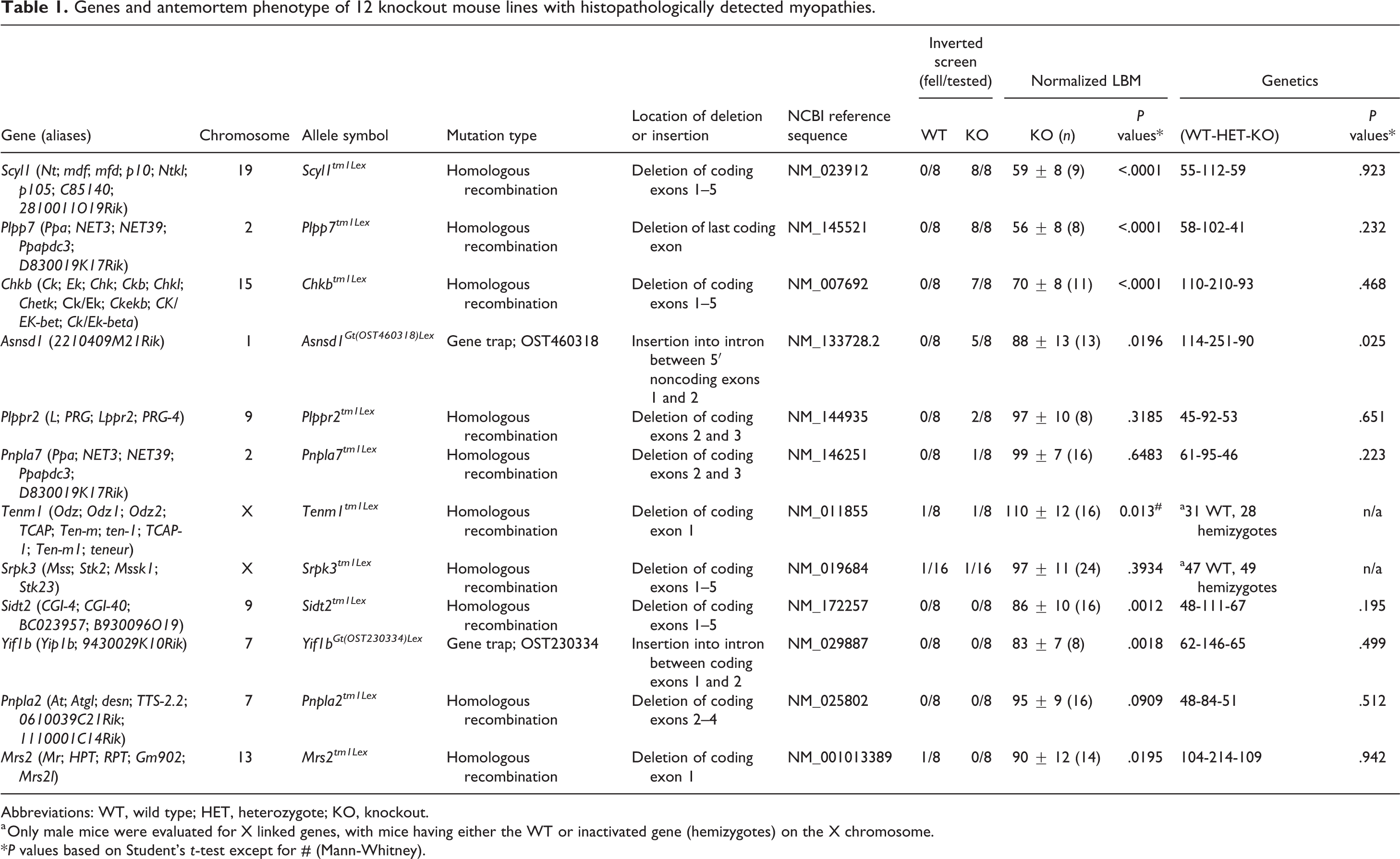
Abbreviations: WT, wild type; HET, heterozygote; KO, knockout.
a Only male mice were evaluated for X linked genes, with mice having either the WT or inactivated gene (hemizygotes) on the X chromosome.
*P values based on Student’s t-test except for # (Mann-Whitney).
For those lines generated by homologous recombination, we utilized both λ phage KOS shuttle systems 57 and PCR-based targeting vector strategies as described previously. 62 Exons of interest were deleted and replaced with a selection cassette containing the neomycin gene. To generate pKOS targeting vectors, the λ phage KOS phage library 57 was screened by duplex PCR using oligonucleotide primers complementary to the gene of interest. The genomic clone identified in this screen was co-transformed into yeast with a URA3 yeast selection cassette that had been appended with gene-specific arms flanking the desired deletion interval. Clones that had undergone homologous recombination with the yeast-selectable marker were isolated, and the yeast cassette was replaced with an ES cell selection cassette containing the reporter gene LacZ and the neomycin gene. The targeted or gene-trap mutations were generated in strain 129SvEvBrd-derived ES cells and chimeric mice were bred to C57BL/6J albino mice to generate F1 heterozygous animals. These F1 progeny were intercrossed to generate the F2 progeny (WT, heterozygous and homozygous) used for high-throughput phenotyping studies.
Mouse Husbandry and Care
Mice were housed in a temperature-controlled environment on a fixed 12-hour light/12-hour dark cycle, with free access to water and food. All experimental protocols were approved by the Institutional Animal Care and Use Committee in strict accordance with criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the US National Academy of Sciences. 35
Pathology
For baseline screening, 2 KO mice (1 male and 1 female) and 2 WT littermates were fixed by cardiac perfusion with 10% neutral buffered formalin. Standard tissues examined included heart, skeletal muscle, aorta, lung, kidney, renal lymph node, trachea, thyroid gland, parathyroid gland, mediastinal lymph node, adrenal gland, pituitary gland, thymus, salivary glands, cervical lymph node, esophagus, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, rectum, mesenteric lymph node, liver, gallbladder, spleen, brain, spinal cord, eyes, Harderian gland, urinary bladder, uterus, ovaries, fallopian tube, skin, mammary gland, bone, bone marrow, adipose tissue, blood, teeth, prostate gland, testes, epididymis, seminal vesicle, vas deferens, urethral glands, inguinal lymph node, inner ear, middle ear, and nasal turbinates. Tissues were collected and immersed in 10% neutral buffered formalin for an additional 48 hours except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 µm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific) and stained with hematoxylin and eosin (H&E) for histopathologic examination.
For advanced pathology phenotyping of known or suspected myopathies, more extensive tissue collection was performed to assess motor neurons, myocytes, dorsal root ganglia, peripheral nerves, neuromuscular junctions, and mechanoreceptors. Because most lesions are more easily detected in transverse sections of both the spinal cord and skeletal muscle, we used at least 6 cassettes for adult mice; 4 of these contained maximal cross-sections of the spinal cord, while the remaining 2 cassettes contained lateral and cross-sections of the rear legs. The lateral orientation included longitudinal sections of skeletal muscle above and below the knee joint. The second cassette contained at least 6 cross-sections of the contralateral rear leg (3 transverse sections above the knee and 3 more below the knee).
Lean Body Mass Analysis
As part of our high-throughput primary screen of KO lines fed standard rodent chow diet (9F 5020; Purina) from weaning, we used dual-energy X-ray absorptiometry (DXA; PIXImus, GE Medical Systems) to analyze the body composition of 3651 KO lines at 14 weeks of age. For each KO line reported here, we calculated the mean KO lean body mass (LBM) divided by the mean WT littermate LBM for both male and female mice. These values for both male and female mice were then pooled, yielding a normalized LBM value, as described previously. 38 As part of our high-throughput primary screen of KO lines fed 45% fat diet (HFD, D12451i; Research Diets) from weaning, we also used quantitative magnetic resonance (QMR; ECHO Medical Systems) to analyze body composition of male mice from 2463 KO lines at 11 weeks of age; for each KO line, mean KO LBM/mean WT littermate LBM was calculated in order to generate a normalized LBM value as described above. Because there is a strong correlation between LBM measured by DXA and by QMR, 39 normalized LBM data from the 2 mouse cohorts were pooled for statistical analysis (Table1).
Inverted Screen
Mice were placed individually on top of a square (7.5 cm × 7.5 cm) wire screen mounted horizontally on a metal rod. The rod was then rotated 180° so that the mice were hanging upside down on the bottom of the screen. Sixty seconds after inverting the screen, the following behavioral responses were recorded and scored as follows: fell off the screen = 0, remained on the bottom of the screen but did not move = 1, remained on the bottom of the screen but moved around = 2, remained on the screen and climbed part way to the top = 3, and climbed to the top of the screen = 4.
Statistical Analysis
Lean body mass (LBM) data are presented as mean ± standard deviation (SD). Comparisons between WT and KO mice were analyzed by unpaired Students t-test, except for comparisons between 2 groups with significantly different variances, which were analyzed by the Mann-Whitney test. Chi-square testing was used to evaluate whether Mendelian ratios deviated from expected values. For all comparisons, differences were considered significant when P < .05.
Results
Scyl1
The inverted screen test demonstrated muscle weakness in Scyl1-/- mice, with 8/8 falling off the screen. In addition, LBM (which is often used as a surrogate for skeletal muscle mass in mammals 9 ) was markedly reduced in the Scyl1-/- mice. Histological lesions were present in 8/8 Scyl1-/- mice and were characterized by neuropathic myopathy/atrophy of skeletal muscle, degeneration and loss of lower motor neurons, and axonal degeneration in peripheral nerves. Widespread changes in affected muscles included extreme variations in muscle fiber diameter, with groups of angulated (atrophied) fibers often adjacent to less severely affected rounded myocytes, reflecting the functional loss of individual lower motor neurons (Fig. 1). No degenerative or regenerative changes were evident in skeletal muscle, and inflammatory cell infiltrates were absent.
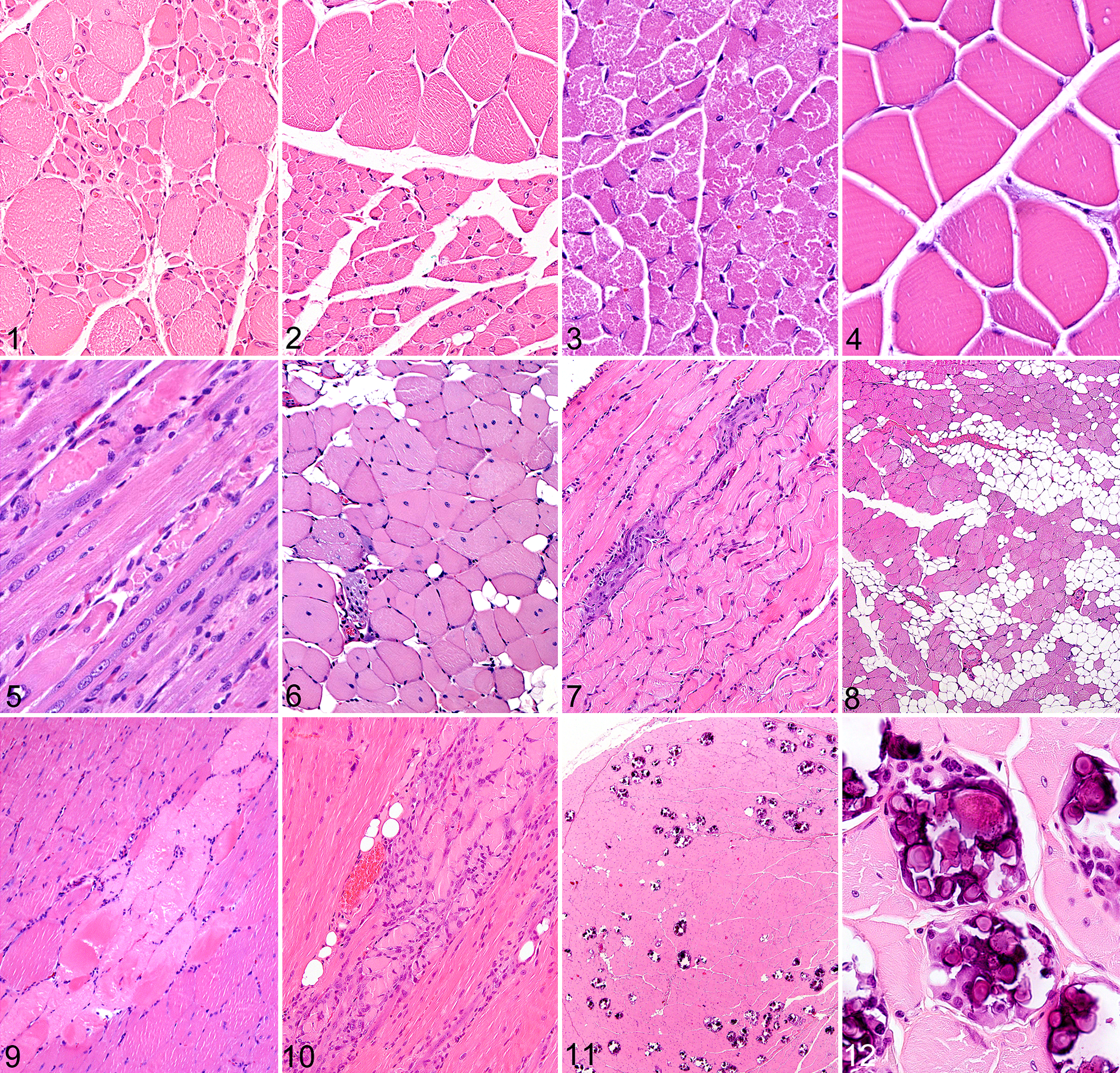
Skeletal muscle, mouse. Hematoxylin and eosin.
Notably, the severity of neuropathic atrophy differed markedly between specific muscles (Fig. 2). For example, in the rear leg, the quadriceps muscle was severely affected, while most posterior muscles of the rear leg showed mild to no evidence of neuropathic atrophy. The muscle fibers in the less affected muscles were noticeably rounder and smaller than those of WT mice, but the normal peripheral localization of nuclei and generally uniform diameter of the rounded myocytes in these fascicles are suggestive of disuse atrophy, secondary to the neuropathic atrophy affecting the anterior muscles. Similarly, in the forelegs, neuropathic atrophy of the anteriorly located biceps brachii muscle was accompanied by diffuse disuse atrophy of the posteriorly located triceps brachii muscle.
In other screening assays, Scyl1-/- mice exhibited increased systolic blood pressure and numerous neurological and immunological abnormalities, and male Scyl1-/- mice were infertile.
Plpp7
Plpp7-/- mice were easily identifiable at weaning age due to stunted growth, which is consistent with their decreased LBM and severe hindlimb weakness. The inverted screen test clearly demonstrated muscle weakness in Plpp7-/- mice, with 8/8 falling off the screen. Multiple behavioral abnormalities associated with weakness/movement defects were detected in the Plpp7-/- mice. Histopathology showed a diffuse marked decrease in muscle fiber diameters in Plpp7-/- mice (Fig. 3) when compared with WT littermates (Fig. 4). The small diameter myocytes were characterized by round rather than polygonal shape on cross-section. In longitudinal sections, muscle fibers were less than half the thickness of those in littermate controls and showed a markedly increased density of myocyte and satellite cell nuclei per unit area. In contrast to the severe diffuse myocyte hypoplasia, peripheral nerves and myelinated tracts in the spinal cord appeared to be entirely normal in appearance and thickness. The numbers and appearance of both upper and lower motor neurons were also normal. Microscopic analysis of 3-week-old (n = 4) and 12-week-old (n = 4) Plpp7-/- mice showed that myopathy was nonprogressive and lacked any histological evidence of muscle fiber degeneration/necrosis or inflammation.
Other phenotyping assays indicated that Plpp7-/- mice had significantly reduced fat mass, reduced cortical and trabecular bone density, elevated blood phosphate levels, and decreased creatinine levels. RBC, hemoglobin, hematocrit, and platelet levels were all increased, and glucose tolerance was enhanced.
Chkb
Chkb-/- mice showed numerous clinical abnormalities, including retarded growth consistent with their decreased LBM, deformed forelimbs, hindlimb weakness, and infertility. The inverted screen test clearly demonstrated muscle weakness in Chkb-/- mice, with 7/8 falling off the screen. The skeletal muscle myopathy was present in 4/4 Chkb-/- mice, and was minimal to absent in all muscles except in the distal rear leg, where the gastrocnemius muscle was severely affected. Lesions in the gastrocnemius muscle were characterized by shrunken fibers, disrupted and coagulated sarcoplasm, centralization and proliferation of nuclei, basophilic regenerative fibers, and minimal fibrosis (Fig. 5). In other muscles, lesions were multifocal and mild (manifesting as prominent mitochondria and irregular myofibrillar striations). Other histologic lesions were present in skin (diffuse hyperkeratosis and sebaceous gland hyperplasia) and organ of Corti (absence of sensory hair cells), but no abnormalities were detected by other phenotyping assays.
Asnsd1
As we previously reported, 49 muscle lesions and increased total body fat were present in 8/8 KO mice evaluated at 2 different time points. The inverted screen test demonstrated the reduced strength of Asnsd1-/- mice, with 5/8 falling off the screen. In addition, LBM was significantly decreased in Asnsd1-/- mice. At the earlier stages (14 weeks), most fibers appeared to be normal although there were scattered individual and small groups of muscle fibers exhibiting features of degeneration such as internalized nuclei (Fig. 6), hypereosinophilia (hyaline degeneration) and small foci of regeneration (Fig. 7). In some locations there were a few necrotic cells with disrupted flocculated sarcoplasm, sometimes containing intracellular macrophages. In older mice (37 weeks) there was extensive internalization of myocyte nuclei and disruption of muscle architecture and degeneration in many locations. 49 Widely scattered hypereosinophilic degenerating fibers contained macrophages, while less severely affected atrophic fibers showed disordered myofibril alignment and disrupted banding patterns. There was extensive replacement of muscle by well-differentiated adipose tissue, and only minimal fibrosis and muscle regeneration were associated with this myopathy (Fig. 8). Other than macrophage infiltrates within and adjacent to necrotic myocytes, inflammatory cell infiltrates were rarely present.
Plppr2
Plppr2-/- mice appeared to be clinically normal; however, histopathology revealed a widespread myopathy in 4/4 Plppr2-/- mice examined. The muscle lesions were characterized by diffuse internalization/centralization of myocyte nuclei, with multifocal degeneration and necrosis of myocytes (Fig. 9). Internalized nuclei were often associated with fiber splitting, myofiber disarray, and coagulation necrosis and infiltrating macrophages (Fig. 10). There were extensive areas with mineralized and necrotic myocytes (Fig. 11), the mineralization being characterized by dense aggregates of basophilic lamellated nodules surrounded by multinucleated foreign-body giant cells (Fig. 12). The more severe lesions were generally accompanied by mild to moderate interstitial fibrosis, and interstitial fibrosis was also present in the heart muscle. The muscle and cardiac lesions tended to be more extensive and severe in male Plppr2-/- mice than in female mice. Other phenotyping assays showed that Plppr2-/- mice tended to have reduced TNFalpha, MCP-1, and IL-6 in the acute phase response assay, as well as decreased blood pressure.
Pnpla7
Histopathologic evaluation of Plpla7-/- mice revealed notable lesions in both skeletal and cardiac muscle in 4/4 mice examined. The myopathy was characterized by widely disseminated centralization of nuclei, degeneration and necrosis of individual skeletal myocytes, and minimal histological evidence of regeneration (Fig. 13). The myopathy was characterized by enlarged nuclei, sometimes having prominent eosinophilic nucleoli (Fig. 14), fiber splitting, circular myofibrils, hypereosinophilic and vacuolated fibers, degeneration with mineralization, and mild inflammation. In addition, mild inflammatory and degenerative lesions were found in several other tissues in Plpla7-/- mice. These lesions were characterized by necrosis of individual cells in exocrine cells (salivary glands and pancreas), liver, and adipose tissue. Pnpla7-/- mice showed elevated total WBC and lymphocyte counts, and increased IL-6 levels, but were essentially normal in all other screening assays except for histopathology. The inflammatory lesions were generally accompanied by inflammatory cell infiltrates, which would account for the elevated WBC counts and IL-6 levels observed in these mice.
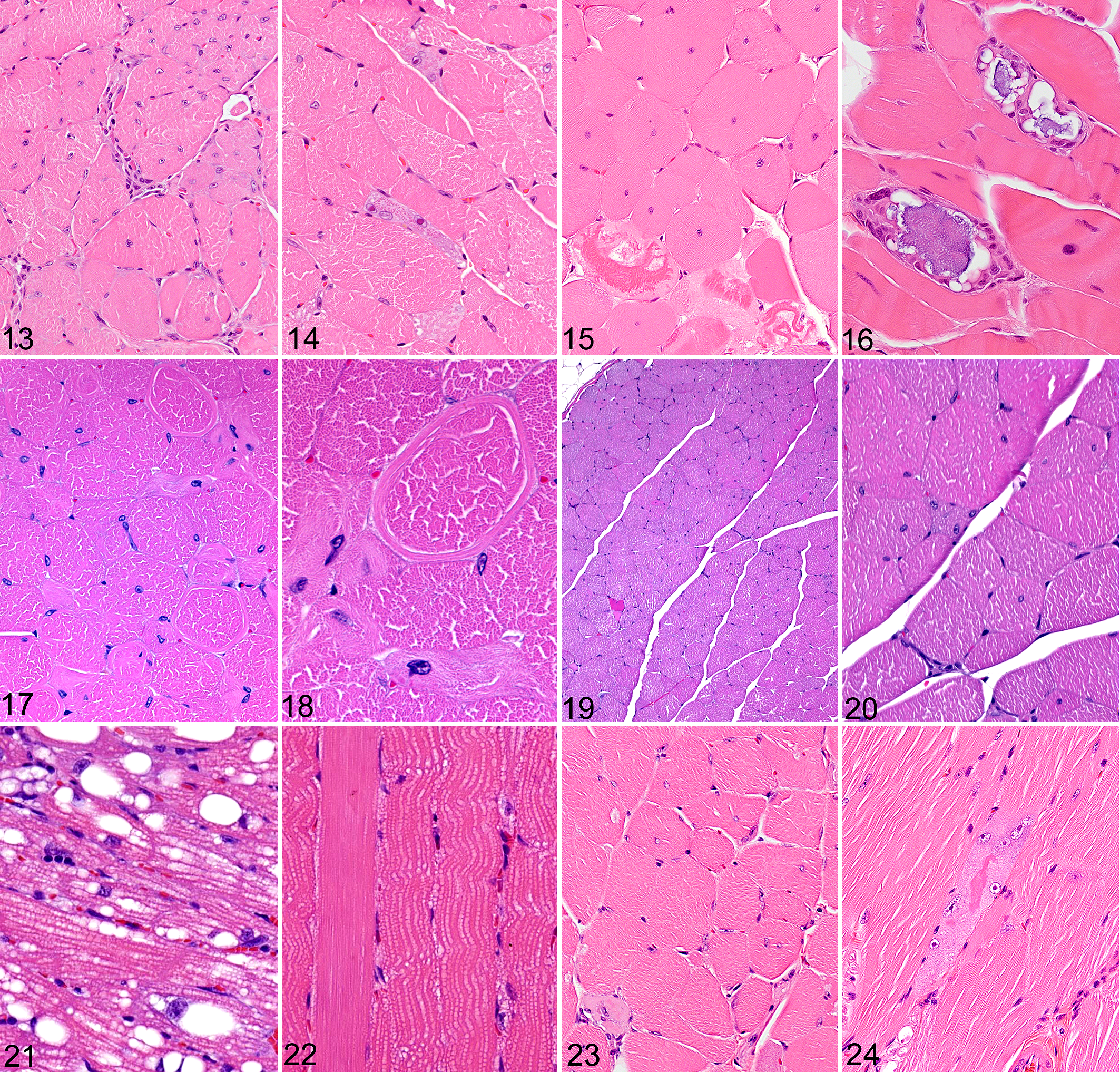
Skeletal muscle, mouse. Hematoxylin and eosin.
Tenm1
Hemizygous (Tenm1-/0 ) mice did not show notably decreased motor strength compared to WT controls in the inverted screen assay, and in contrast to most other KO lines reported here, the LBM was significantly increased rather than being decreased. Nevertheless, lesions were detected in the skeletal muscle of 15/15 male Tenm1-/0 mice evaluated by histopathology. The myopathy in these mice was characterized by extensive internalization of myocyte nuclei, abundant rounded myofibers of varied size, and scattered degenerating and necrotic myofibers (Fig. 15). The mineralization in these myocytes consisted of coarse basophilic material that was surrounded by multinucleated giant cells (Fig. 16). The only other phenotype detected in hemizygous mice by baseline screens was an increased serum IgG2a response to ovalbumin challenge.
Srpk3
Hemizygous (Srpk30/- ) mice developed normally and showed no clinical signs of muscle weakness. Histologically, 4/4 male Srpk30/- mice displayed a nonprogressive, minimal to moderate, centronuclear myopathy (Fig. 17), with occasional myofibril disarray (ring fibers and swirls; Fig. 18). As in the previously reported line of mice, 34 the muscle lesions were restricted to type II muscle fibers, and lacked any histologic evidence of advanced myocyte degeneration/necrosis or inflammation. The only other phenotypes detected in hemizygous mice by baseline screens were decreased serum IgG1 and IgG2a responses to ovalbumin challenge and increased tear production.
Sidt2
Sidt2-/- mice did not show decreased motor strength compared to WT controls in the inverted screen assay but did show a significant decrease in LBM. We observed multifocal mild to moderate degenerative myopathy in 6/6 Sidt2-/- mice. The widely scattered skeletal muscle lesions were present within individual and small clusters of myocytes. The degenerative lesions in myocytes were characterized by centralization of nuclei, eosinophilic/coagulated sarcoplasm, loss of striations, vacuolization, and shrunken necrotic fibers. An inflammatory component consisting of infiltrating granulocytes and macrophages was usually associated with areas of coagulative necrosis involving individual myofibers. In addition, there were some foci with regeneration (small basophilic fibers contain rows of large centralized nuclei). The only other phenotype detected in Sidt2-/- mice was a decreased serum IgG1 response to ovalbumin challenge. 27
Yif1b
Yif1b-/- mice were significantly smaller than WT littermates consistent with their significantly decreased LBM, and also exhibited enhanced glucose tolerance and a decreased pain response.
Yif1b-/- mice did not show decreased motor strength compared to WT controls in the inverted screen assay. Histopathology revealed a mild myopathy in 4/4 Yif1b-/- mice. The myopathy was characterized by widespread centralization of myocyte nuclei, with rare degenerating/necrotic myocytes (Fig. 19) and small clusters of atrophic fibers with centralized nuclei (Fig. 20). Other notable histopathological lesions were restricted to cerebellum and spinal cord and included mild cerebellar hypoplasia and small foci containing degenerating neurons, spheroids, gliosis, and mild vacuolization of surrounding neuropil in the deep cerebellar nuclei and spinal cord.
Pnpla2
Pnpla2-/- mice showed variably sized lipid droplets in many different tissues, including the heart and skeletal muscle, in 4/4 mice examined. Lipid accumulation was much more severe in the heart (Fig. 21) than in skeletal muscle (Fig. 22), but swollen fibers and disruption of myofibrils were evident in both tissues. In other screening assays, Pnpla2-/- mice exhibited elevated IL-6 and TNF levels in the acute phase response, increased serum creatinine, reduced depressive-like response, increased anxiety-like behavior, and reduced sensitivity to tonic pain.
Mrs2
Mrs2-/- mice did not show decreased motor strength compared to WT controls in the inverted screen assay but did show a significant decrease in LBM. Skeletal muscle lesions were detected in 9/12 Mrs2-/- mice examined. When present, the muscle lesions were generally mild, being characterized by variation in fiber size and centralization of nuclei (Fig. 23). Degenerative changes were present but rare, and inflammation was notably absent (Fig. 24). Most skeletal muscle lesions were located within the rectus femoris muscle and were minimal to absent in other muscle groups. The predominant histologic lesions in Mrs2-/- mice were localized to adipose tissue. While brown fat in WT mice consists of generally uniform multilocular adipocytes containing myriad small lipid droplets, the size and appearance of BAT adipocytes in Mrs2-/- mice varied tremendously, with small numbers of normal brown adipocytes being surrounded mostly by adipocytes distended by a single large lipid vacuole as well as adipocytes containing abundant eosinophilic cytoplasm and minimal lipid. Altered adipocyte morphology was also widespread in abdominal/visceral white adipose tissue, where the unilocular white adipocytes were reduced in size and more than half had converted into multilocular adipocytes. In contrast, the subcutaneous white adipocytes retained a more normal unilocular cytoplasm.
Discussion
It is clear that including histopathology in baseline high-throughput phenotyping programs will uncover disease phenotypes that are not detected by any other methods. 2,44 Our findings indicate that histopathologic evaluation of skeletal muscle is required for the detection of many myopathies. Mice with advanced or severe myopathies may be stunted, shaky, weak, or display postural or gait abnormalities, and these clinical signs of neuromuscular disease can be confirmed using behavioral assays such as the inverted screen or grip strength tests. However, mild signs are not always clinically evident, particularly in younger mice.
The inverted screen test is the most commonly used method used to assess skeletal muscle strength in high-throughput screening assay, but it detected muscle weakness in only 4 of the 12 KO mouse lines (Scyl1, Plpp7, Chkb, and Asnsd1) in which we found significant myopathies. Given the clinical signs and inverted screen test results it is not surprising that the myopathies in these 4 lines have been recognized and published previously (Scyl1 and Asnsd1, by authors of this report). In contrast, myopathies have been recognized and reported in only 2 (Srpk3 and Sidt2) of the 8 other KO mouse lines reported here, all of which showed essentially normal results on the inverted screen test. Because skeletal muscle constitutes ∼2/3 of LBM in healthy mammals, 9 the skeletal muscle wasting associated with some myopathies can manifest as low LBM. However, the diagnostic specificity and sensitivity of using LBM to detect myopathies is poor, mostly because there are many other disease processes can result in reduced LBM, and also because we show here that 4/12 KO mouse lines with myopathies had normal or even increased LBM.
Our findings indicate that histopathology must be included in phenotype screening protocols in order to identify novel genetic myopathies that are not clinically evident or detectable by the inverted screen test. Histopathologic evaluation of transverse sections of the spinal column and legs can detect and characterize early or subclinical myopathies that cannot be demonstrated with commonly used strength and behavioral screening tests. Histologic findings can also provide critical information regarding the likely pathogenesis of the myopathy.
Myopathies can be broadly classified as either neuropathic or myogenic, based on the primary underlying pathogenic mechanisms responsible. In our experience with neuropathic disease, the severity of atrophy can vary widely between different muscle groups, with distal muscles are often affected earlier in the disease process. In these cases, affected myocytes are generally small, appear in clusters, and are adjacent to normal or hypertrophic polygonal myocytes. We have not seen myofiber degeneration and necrosis, with accompanying inflammatory cell infiltrates, associated with neuropathic myopathies. An additional sign we have often observed in neuropathic myopathies is that most cell nuclei remain at the periphery of the atrophic fibers, in contrast to the internalized nuclei typically found in myogenic myopathies. We observed the presence of groups of small angular fibers in affected muscles, sometimes with fiber type conversion, in cases of chronic neuropathic atrophy due to cycles of denervation and reinnervation.
Since the myogenic myopathies are caused by defects intrinsic to myocytes, they generally have a more diffuse distribution in skeletal muscle than the neuropathicmyopathies. Histologically, we have found internalization of myocyte nuclei to be an extremely sensitive early indicator of a myogenic myopathy. The internalized nuclei are more easily detected in muscle cross-sections than in longitudinal sections, where they are generally missing in most myocytes due to the plane-of-section effects. Other common findings associated with myogenic myopathies include myofibril disarray, degeneration and necrosis of myofibers, coagulation and fragmentation of sarcoplasm, and inflammatory infiltrates. Degenerating fibers can also appear in clusters with myogenic myopathies, but in contrast to the peripheral nuclei seen with neuropathic myopathies, the nuclei in myogenic myopathies are generally centralized or absent. Mineralization, fibrosis, and inflammation are additional common findings in many chronic myogenic myopathies.
There are also mixed neuropathic/myogenic myopathies, which are caused by dysfunctional cellular processes critical to both neurons and myocytes (such as autophagy). Neuropathic, myogenic, and mixed myopathies can all result in decreased muscle mass due to myocyte atrophy and/or loss. By supplementing routine hematoxylin and eosin staining of muscle with appropriate immunohistochemical labeling, a pathologist can usually distinguish between neuropathic or myogenic origins of neuromuscular symptoms.
Scyl1 (SCY1-like 1 [S. cerevisiae])
Scyl1 is a highly conserved and ubiquitously expressed gene encoding the COPI-associated protein pseudokinase SCYL1. Findings in our KO mice were essentially identical to those we reported in another line of Scyl1-/- mice. 36 Clinically, mice showed severe growth retardation and motor dysfunction characterized by hindlimb paralysis. Pathologic findings included neuropathic myopathy/atrophy of skeletal muscle, degeneration and loss of lower motor neurons, and axonal degeneration in peripheral nerves. A later study showed that neuron-specific deletion (but not skeletal muscle-specific deletion) of Scyl1 was sufficient to cause motor dysfunction, indicating that SCYL1 is required to prevent lower motor neuron degeneration but that it is not essential for maintaining skeletal muscle homeostasis. 36
Plpp7 (Phospholipid Phosphatase 7 [Inactive])
PLPP7 is a muscle-specific nuclear envelope transmembrane protein (NET) that has no detectable enzymatic activity. However, PLPP7 is one of several NETs (including Tmem38A, WFS1, Tmem214, and NET5/Samp1) whose function is to move genes to the nuclear periphery where they are essentially shut down. 26 This NET-directed gene repositioning is critical for myocyte differentiation, 41,67 and in mouse cell culture, Pllp7 is upregulated over 40-fold, where it appears to control MTOR-dependent IGF2 expression during myoblast differentiation. 26 Many of the genes downregulated by muscle-specific NETs encode proteins that block myotube formation, which is the process by which relatively undifferentiated cells acquire the specialized characteristics of multinucleated myocytes. Therefore, loss of muscle-specific NETs (including PLPP7, Tmem38A, WFS1, and NET5/Samp1) results in those genes being de-repressed, and their proteins blocking myotube fusion. 41
The hypoplastic myopathy we observed in Plpp7-/- mice was not entirely surprising given that other NETs have been linked to the development of Emery-Dreifuss muscular dystrophy (EDMD) in humans, 60 and PLPP7 is specifically downregulated in EDMD, 30 which presents as a slowly progressive genetic disorder affecting the muscles of the arms, legs, face, neck, spine, and heart. Our histologic findings were indicative of a nondegenerative primary developmental defect affecting all myocytes. Although there is clear evidence that PLPP7 is required for differentiation of muscle, and other lines of Plpp7-/- mice have been produced, we are not aware of any previous publications showing the histologic lesions due to PLPP7 deficiency in mice.
Chkb (Choline Kinase Beta)
This choline kinase catalyzes the phosphorylation of choline to phosphocholine in the biosynthesis of the major membrane phospholipid phosphatidylcholine. Choline kinase in mice is encoded by 2 genes, Chka and Chkb. Disruption of murine Chka leads to embryonic lethality, whereas a spontaneously occurring genomic deletion in murine Chkb results in neonatal bone deformity and a hindlimb muscular dystrophy. 46
These muscle and forelimb lesions were virtually identical to those reported previously in CHKB deficient mice (Chkbrmd ) that developed a progressive muscular dystrophy with a rostral-to-caudal gradient of severity, accompanied by forelimb bone deformities. The spontaneous recessive mutation in those mice is a 1.6-kb intragenic deletion within the choline kinase beta (Chkb) gene, resulting in a complete loss of CHKB protein and enzymatic activity. 46
Yet another line of Chkb-/- mice also developed neonatal forelimb bone deformity and hindlimb muscular dystrophy (with sparing of the forelimb musculature). The mitochondria in these Chkb-/- mice were abnormally large and exhibited decreased inner membrane potentials. The muscular dystrophy in these Chkb-/- mice was attributed to reduced phosphocholine biosynthesis and increased phosphocholine catabolism in the hindlimbs. 59 Later, the same group determined that choline kinase beta is the major isoform in the susceptible hindlimb muscles whereas choline kinase alpha is predominant in the largely unaffected forelimb muscles. 58
After the identification of CHKB-related muscular dystrophy in mice, a congenital muscular dystrophy in humans characterized by early-onset muscle wasting, mental retardation, and enlarged mitochondria was linked to homozygous or compound heterozygous mutations in the gene encoding choline kinase beta (CHKB). 31
Asnsd1 (Asparagine Synthetase Domain Containing 1)
The gene encoding asparagine synthetase domain containing 1 (ASNSD1) is conserved across many species, and whole-body gene expression surveys show that ASNSD1 is highly expressed in skeletal muscle and many other tissues, including adipose tissue (https://gtexportal.org/home/). However, potential functions of this protein had not been previously reported until we showed that mice with an inactivating mutation in Asnsd1 develop a progressive degenerative myopathy that results in severe sarcopenia and myosteatosis. Histologic lesions were essentially limited to the muscle and were characterized by a progressive degenerative myopathy. 49
Plppr2 (Phospholipid Phosphatase Related 2)
The lipid phosphate phosphatase-related proteins (LPPRs), also known as plasticity-related genes (PRGs), comprise a family of transmembrane proteins that are enriched in brain, with 5 members (LPPR1–LPPR5) having been identified to date. 47 The function of PLPPR2 is still unknown, although it is predicted to be localized to the plasma membrane and be involved in signal transduction via the dephosphorylation of phospholipids.
Pnpla7 (Phospholipid Phosphatase 7 [Inactive])
The patatin-like phospholipase domain–containing proteins (PNPLAs) are enzymes that have essential roles in lipid metabolism. PNPLA7 is a transmembrane protein that specifically promotes hydrolysis of lysophosphatidylcholine. 20 Although the in vivo functions of PNPLA7 are still largely unknown, 33 a probable role in maintaining metabolic homeostasis is suggested by its direct catalytic interaction with lipid droplets in cells 20 and by its downregulation by insulin in white adipose tissue. 23
The association of PLPLA7 deficiency with skeletal muscle lesions has not been reported previously, but deficiencies of other PNPLAs have been linked to the development of myopathies. For example, Pnpla8-/- mice displayed signs of metabolic and neuronal dysfunction, leading to muscle weakness and atrophy. 28 Humans with loss-of-function mutations in PNPLA8 gene show similar neuromuscular signs which were attributed to mitochondrial dysfunction related to defective lipid metabolism. 43 Mutations in PNPLA2, which encodes an adipose triglyceride lipase, are associated with a lipid storage disease characterized by the presence of cytoplasmic lipid droplets in many tissues, including skeletal muscle. 14,22,40
Tenm1 (Teneurin Transmembrane Protein 1)
Teneurins are highly conserved large transmembrane proteins first identified in Drosophila melanogaster 6 where they are required for organizing neuromuscular synapses. 32 In mammals, Tenm1 is an X-linked gene involved in synapse organization in the olfactory system of mice, and inactivating mutations in TENM1 have been linked to congenital general anosmia in humans. 3 Tenm1-/- mice and Tenm1-knockin mice showed no visible phenotype or differences in their locomotor activity compared to Tenm1+/+ mice, but no histological assessments were completed or reported. 3 However, both Tenm1-/- mice and Tenm1-knockin mice showed impaired odor discrimination ability when tested against the aversive odorant 2-methyl butyric acid, and they took significantly longer to locate a buried food pellet compared with WT mice. 3 Although TENM1 has not been directly associated with myopathy in humans, this gene was one of 21 associated with myopathy in a genome-wide association study. 42
Srpk3 (Serine/Arginine-Rich Protein Specific Kinase 3)
Srpk3 is an X-linked gene that encodes a protein kinase that is specifically expressed in the heart and skeletal muscle and involved in the development and homeostasis of skeletal muscle throughout life. 34 It is known that Srpk3 is directly regulated by myocyte enhancer factor 2 (MEF2), which plays essential roles in the transcriptional control of cardiac and skeletal muscle development and remodeling. 34 Significantly, Srpk3 expression is downregulated in poorly differentiated rhabdomyosarcoma cells whereas increased expression of SRPK3 promotes alternative splicing of an MEF2Cα2 isoform that induces myocyte differentiation. 65 Increased expression of either MEF2Cα2 or SPRK3 inhibits both the proliferation and anchorage-independent growth of rhabdomyosarcoma cells, which indicates that the SPRK3-mediated alternative splicing of MEF2C may have an important role in normal myogenesis and in reducing rhabdomyosarcoma development. 65 Our findings were very similar to those reported previously in another line of Srpk3 0/- mice, which also displayed a type 2 fiber-specific myopathy characterized by increased centralized nuclei but no evidence of myocyte degeneration and regeneration, such as muscle cell death, myofiber disarray, or inflammation. 34 The relatively mild nonprogressive muscle phenotype in both lines of Srpk3 0/- mice suggests that SRPK3 has a peripheral role in myocyte differentiation and maintenance.
Sidt2 (SID1 Transmembrane Family, Member 2)
SIDT2 is a lysosomal membrane protein involved in translocating substrate proteins. 17
We did not observe the severe steatohepatitis reported in another line of Sidt2-/- mice. 17 Those Sidt2-/- mice also exhibited a remarkable accumulation of the classical autophagy pathway marker proteins (p62 and LC3-II) in hepatocytes, suggesting that Sidt2 deficiency disrupts hepatic lipid homeostasis by blocking autophagosome maturation. 11 Recent findings confirm that defects in the lysosomal autophagic pathway may contribute to the pathogenesis of NAFLD by decreasing fatty acid oxidation. 11 A skeletal muscle-specific SIDT2-deficient mouse model developed lesions characteristic of autophagic vacuolar myopathy, 27 which included loss of muscle mass, muscle weakness, fibrosis, centralized nuclei, and fiber regeneration. In addition to an accumulation of autophagolysosomes, there were significant increases in LC3-II, adaptor protein p62, ubiquitinated aggregates, and Lamp2-positive vacuoles in Sidt2 f/fCre skeletal muscle fibers. The relevance of Sidt2-/- mice as models of autophagy myopathy was strengthened by recent findings showing that loss-of-function mutations in SIDT2 also cause a vacuolar myopathy in humans. 16
Yif1b (Yip1 Interacting Factor Homolog b [S. cerevisiae])
Yif1B is a highly conserved intracellular membrane-bound protein belonging to the Yip family. It is present in 2 isoforms in mammals: Yip1A is the ubiquitous isoform while Yip1B is the muscle-specific isoform. 48 The proposed functions of YIF1B include organizing the endoplasmic reticulum and Golgi apparatus as well as traffic from the endoplasmic reticulum to the cell membrane. 12,13 Yif1B influences anterograde traffic in HeLa cells and neurons and is involved in the maintenance of Golgi structure. The Golgi architecture is reportedly disorganized in CA1 pyramidal hippocampal neurons of Yif1B-/- mice. 4
The central nervous system lesions in our Yif1b-/- mice are compatible with recently reported findings in humans and KO mice. Humans with mutations affecting the YIF1B gene present with global developmental delay, motor and locomotor deficits, visual deficits, brain ventricle enlargement, myelination alterations, and cerebellar atrophy. Similar findings were observed in Yif1b-/- mice which displayed neuronal reduction, altered myelination of the motor cortex, cerebellar atrophy, enlargement of the ventricles, and subcellular alterations of endoplasmic reticulum and Golgi apparatus compartments. However, muscle pathology was not reported in either the human patients or KO mice. 12
Nevertheless, it is not entirely surprising to find muscle pathology given that both Yip1 isoforms were shown to be involved in regulating skeletal muscle development. 5 The Yip1A isoform is preferentially expressed in the early stages of muscle development, while Yip1B is the only isoform expressed in adulthood. 5 Both the endoplasmic reticulum and Golgi complex undergo a reorganization that completely remodels the secretory pathway during differentiation of skeletal muscle, with the endoplasmic reticulum being converted into the specialized junctional and longitudinal regions of sarcoplasmic reticulum, which mediate calcium release and uptake during contraction, respectively (reviewed in Barone et al 5 ). In vitro and in vivo knockdown experiments do not alter the structure of the Golgi complex, suggesting that Yip1B is not involved in Golgi complex structure maintenance in skeletal muscle. However, the Yip1B isoform is localized in the endoplasmic reticulum-Golgi intermediate and cis-Golgi compartments in adult skeletal muscle, so long-term effects on myocytes would not be completely unexpected with permanent loss of function in KO mice. 5
Pnpla2 (Patatin-like Phospholipase Domain Containing 2 [Mus Musculus (House Mouse)])
In humans, mutations in the orthologous patatin-like phospholipase domain-containing protein 2 gene (PNPLA2) result in neutral lipid storage disease with myopathy. 10,14 Affected patients present with prominent shoulder girdle weakness and mild pelvic girdle and distal muscle weakness, with cardiomyopathy developing at later stages of disease. 40 Several previous reports have described the effects of inactivating Pnpla2 in mice. Pnpla2 deficiency in mice severely disrupts mitochondrial substrate oxidation and respiration, which leads to excessive lipid accumulation, cardiac insufficiency, and lethal cardiomyopathy. 19 Conditional deletion of Pnpla2 in heart and skeletal muscle was reported to cause a cardiac phenotype only, suggesting that PNPLA2 has a more important role in cardiac muscle than in skeletal muscle. 18
Mrs2 (MRS2 Magnesium Transporter)
Mitochondrial RNA splicing protein 2 (MRS2/MRS2p) is an ubiquitous transporter protein localized to the mitochondrial inner membrane where it mediates the influx of Mg2+ into the mitochondrial matrix. 66 It is required for normal expression of the mitochondrial respiratory complex I subunits. 37 MRS2 overexpression enhances mitochondrial Mg2+ influx whereas mitochondrial Mg2+ influx is abolished with MRS2 gene deletion. 24 The MRS2 protein is therefore described as an essential magnesium transporter in the mitochondria. Cellular energy production processes involve many Mg2+ dependent enzymatic reactions, and dysregulation of Mg2+ homeostasis is involved in various cellular malfunctions and diseases. Rats with functional inactivation of MRS2 have major mitochondrial deficits with a reduction in ATP, and increased numbers of mitochondria in oligodendrocytes, and results in demyelination, 25 but muscle lesions were not reported in these rats.
Normal brown adipocytes are multilocular because they contain many small lipid droplets (multilocular) and abundant mitochondria distributed throughout the cytoplasm. In contrast, white adipocytes have single large lipid droplet (unilocular) with relatively few mitochondria at the cell periphery. The changes in brown and white adipocytes of Mrs2-/- mice most likely are the direct result of altered Mg2+ levels within mitochondria. The conversion of white adipocytes into brown adipocyte-like cells, termed beige adipocytes, can be induced by sustained cold exposure or direct β-adrenergic activation. 21 The WAT-resident beige adipocytes are phenotypically similar to the classical brown adipocytes in BAT, having multilocular lipid droplets and abundant mitochondria expressing uncoupling protein 1 (UCP1), which is present exclusively on the inner mitochondrial membrane of brown adipocytes where it uncouples the respiratory chain from ATP generation. 21
Myopathies are frequently accompanied by lesions in spinal motor neurons, myelinated nerves, motor end plates, or the muscles themselves that are visible by light microscopy. The histopathologic identification of the underlying cause of myopathies in mice depends on the proper fixation and collection of target tissues. For comprehensive neuromuscular disease phenotyping, we recommend whole body transcardial perfusion fixation with 10% neutral buffered formalin followed by paraffin-embedding (FFPE) is essential in order to consistently identify many critical lesions associated with neuromuscular disease. For example, when the spinal cord is fixed by immersion, artifactual cytoplasmic vacuolization and loss of nuclear and cytoplasmic detail will obscure lesions in the nuclei and/or cytoplasm of spinal motor neurons that could be preserved by rapid perfusion fixation. Similarly, many lesions in peripheral nerves, motor endplates and skeletal muscle are not detectable in immersion fixed tissues.
A complete histopathologic examination of spinal cord and skeletal muscle is critically important in uncovering and understanding the etiopathogenesis of novel myopathies. For this, we recommend the collection of multiple cross-sections of the spinal cord and limbs. Inclusion of littermate controls (WT age- and sex-matched) is extremely important for baseline studies. For lines where muscle phenotypes are very mild, aging or challenge (treadmill, wheels) studies should be considered. To evaluate disease progression, tissues should be collected from weaned (3–4 weeks old), mature (3–4 months old), and geriatric (1–2+ years) mice.
In summary, our findings demonstrate that histopathology is a very sensitive and specific method for discovering myopathies that are not reliably detected by other means. These studies also support the need to include histopathology in phenotype screening protocols so that potentially important phenotypes in skeletal muscle (and other organ systems) are not overlooked. In addition, histopathology can provide critical insights into the most likely underlying pathogenetic mechanisms (whether of neuropathic, myogenic, or developmental origin) involved in the development of any newly identified myopathies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.