
Abstract
During meat inspections in pigs, dystrophinopathies are among the muscle lesions targeted for disposal. In this study, the authors examined the lesions and the distribution of dystrophin expression in 25 pigs with dystrophinopathy. In addition, complementary deoxyribonucleic acid (cDNA) sequencing and western blotting were performed in 6 of the 25 cases, all of which were characterized by degeneration, necrosis, and fat replacement of muscle fibers. Comparing the results of immunohistochemistry with anti-dystrophin antibodies that recognized at different sites in the protein, the authors noted that the loss of dystrophin expression was most pronounced in the C-terminus-recognizing antibody (19/25 cases). The authors detected 5 missense mutations and 3 types of shortened transcripts generated by the skipping of exons in the cDNA, which were associated with the pathogenesis. One missense mutation had been reported previously, whereas the remaining mutations identified had not been previously documented in pigs. In the cases with shortened transcripts, normal-sized transcripts were detected together with the defective transcripts, suggesting that these mutations were caused by splicing abnormalities. In addition, they were in-frame mutations, all of which have similar pathogeneses of Becker muscular dystrophy in humans. These cases were 6 months of age and exhibited macroscopic discoloration, fatty replacement, and muscle degeneration, suggesting that the effect of these mutations on skeletal muscle was significant.
Keywords
Muscle lesions, known as muscular steatosis or fatty muscular dystrophy, which are characterized by fatty replacement and pale discoloration of muscles are detected in pigs during meat inspection.17,25,29 Among these degenerative lesions, myopathy with reduced dystrophin expression has been reported. 17
In pigs, dystrophin is a 425 kDa protein that localizes to the sarcolemma.10,37 It connects the cytoskeleton, sarcolemma, and extracellular matrix, and is involved in the structural stabilization of muscle cells.19,39 The dystrophin gene is located on the X chromosome and consists of approximately 11 kb of cDNA comprising 79 exons in pigs. 26 Mutations in dystrophin cause X-linked recessive myopathy in humans and in various other animals.7,35 Dystrophinopathies include Duchenne muscular dystrophy (DMD), which is fatal because of the depletion of dystrophin, and Becker muscular dystrophy (BMD), which is relatively mild and is caused by partial dystrophin loss. 18
In human dystrophinopathies, approximately 60% of patients carry a deletion, 10% have a duplication, and the remaining patients have missense, nonsense, or splicing alterations caused by point mutations in the dystrophin gene.1,18,21 The mutated sites are diverse, and the extent of the lesion for most mutations can be explained by the reading frame hypothesis.18,24 In DMD, the displacement causes a termination codon, resulting in protein instability and, hence, the absence of the dystrophin protein.18,24 Conversely, in BMD, there is no shift in the reading frame, and a shortened or elongated dystrophin is translated.18,24
We have reported 2 cases of dystrophinopathies in pigs detected at meat inspection centers in Japan.3,17 Both cases were diagnosed as BMD-like myopathy because dystrophin expression was reduced but partially retained. Dystrophinopathies in pigs have been reported in Japan, the United States,15,25,30 and Australia. 33 Although reports of dystrophinopathies are limited, cases of muscle steatosis or fatty muscular dystrophy in slaughterhouses are commonly observed. This suggests that dystrophinopathies are a genetic disease and that pigs with dystrophin gene mutations may exist worldwide. In pigs, only 2 mutations in this gene have been reported: a point mutation in exon 41 in a specific strain15,30 and a single case of an insertion of 62 bases in intron 26, as a pseudoexon. 3 The diversity of dystrophin mutations has not been investigated in pigs; therefore, the analysis of additional cases is warranted.
In this study, we examined 25 pigs that were diagnosed with dystrophinopathy at the pathological, protein, and genetic levels to assess the diversity of mutations in the dystrophin gene and its involvement in the pathogenesis of this disease.
Materials and Methods
Cases and Samples
We collected 28 degenerative muscle specimens sent to the university between 2012 and 2022. Twenty-five porcine cases with a diagnosis of dystrophinopathy were examined in the study. The diagnosis of dystrophinopathy was based on the presence of gross findings (skeletal muscle discoloration and fatty replacement), histological findings (degeneration, necrosis, and regenerative changes in myofibers), and the attenuation of the immunohistochemistry (IHC) for dystrophin. Specimens were provided by 3 institutions located in different regions in Japan. Carcass examination was performed in 20 of 25 cases at meat inspection. The distribution of muscular lesions varied, ranging from the gracilis muscles to systemic skeletal muscles (Fig. 1a, b). The details of the cases analyzed here are provided in Table 1. Six out of the 25 cases (4 males and 2 females) from which frozen samples could be procured, underwent genetic/protein examination in addition to pathological investigation. Frozen samples were collected in sealed containers, transported at −20°C, and immediately stored at −80°C.
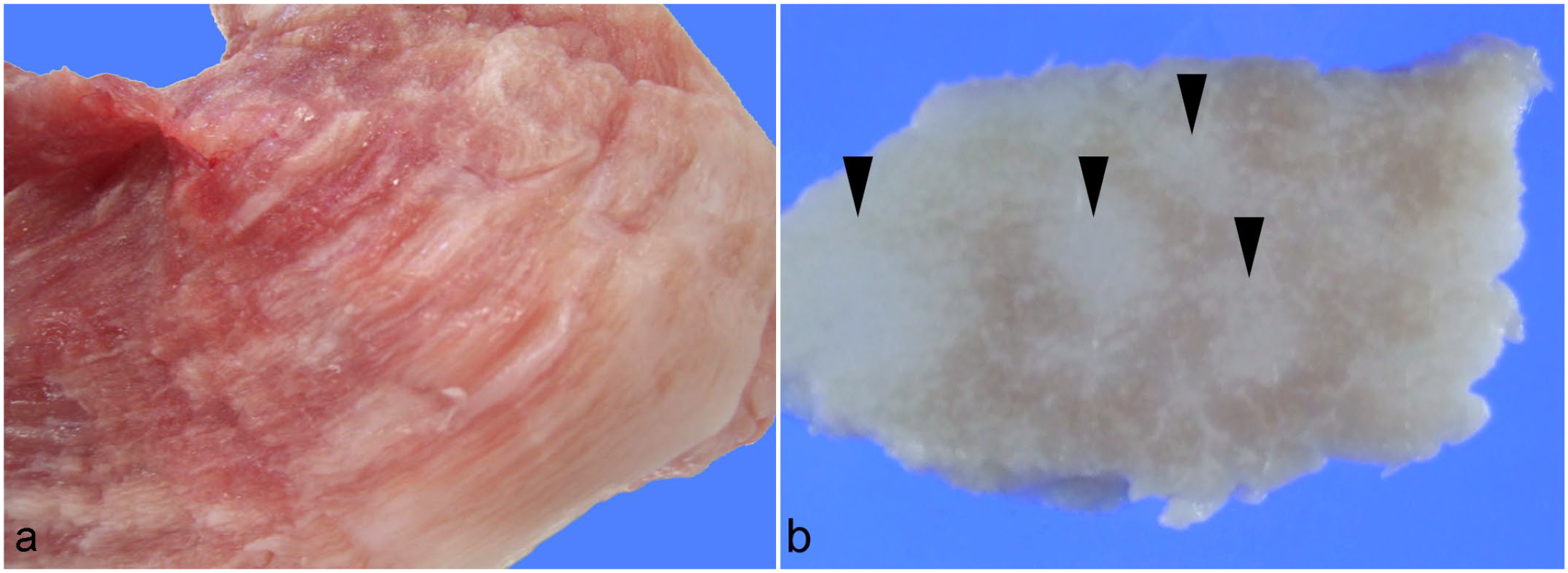
Pale discoloration and fatty replacement in the gracilis muscle of a pig. (a) Poorly circumscribed discoloration in a fresh sample at right side of the figure, case 7. (b) Massive fatty replacement (arrowhead) in a transversal section of muscle after fixation, case 7.
Summary of the case information.
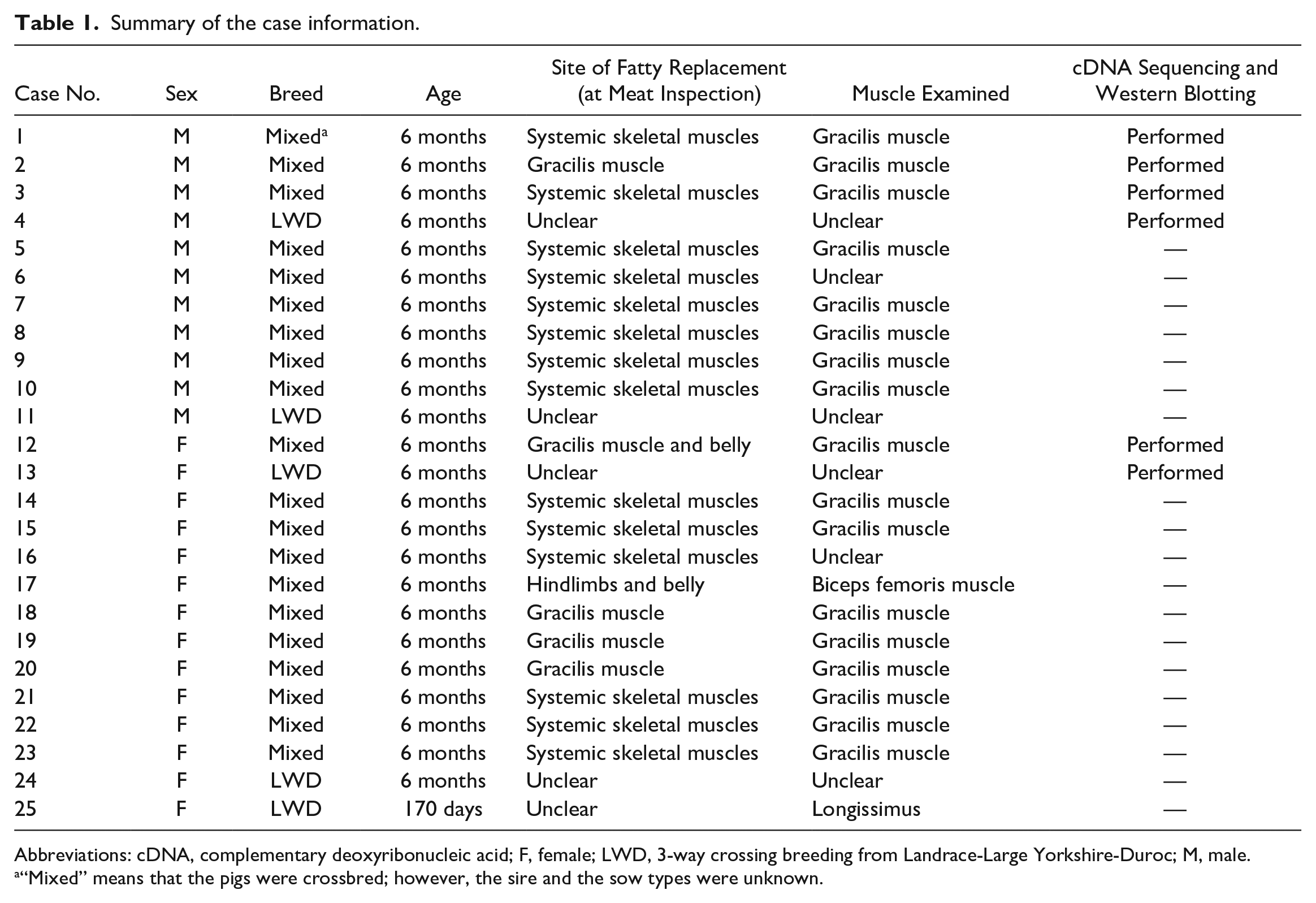
Abbreviations: cDNA, complementary deoxyribonucleic acid; F, female; LWD, 3-way crossing breeding from Landrace-Large Yorkshire-Duroc; M, male.
“Mixed” means that the pigs were crossbred; however, the sire and the sow types were unknown.
As normal controls, gracilis muscles from two 3-month-old male pigs were used. These animals were confirmed to show no gross lesions in the systemic skeletal muscles, no histological changes, and positive with IHC for dystrophin in the gracilis muscles.
Dystrophin Gene Sequencing Analysis
Total RNA was extracted from frozen samples using the TRIzol Reagent (Thermo Fisher Scientific, Waltham, Massachusetts). We used the total extracted RNA for cDNA synthesis using a SuperScript IV First-Strand Synthesis System (Thermo Fisher Scientific). Polymerase chain reaction (PCR) was performed using KOD Fx neo (TOYOBO, Osaka, Japan) according to the primer sets and reaction conditions reported by Aihara et al. 3 The amplified PCR products were purified using NucleoSpin Gel and PCR Cleanup (TAKARA, Shiga, Japan), and their sequences were determined as follows.
The sequencing analysis was performed using MEGA11. The reference sequence was the porcine dystrophin mRNA from the National Center for Biotechnology Information (NM_001012408.1). 26 The impact of the detected mutations on pathology was examined using SIFT (sorting intolerant from tolerant; SIFT: prediction of the effects of nonsynonymous/missense variants)4,27 and PolyPhen-2 (PolyPhen-2: prediction of the functional effects of human nsSNPs).2,14 We selected the mutations with a SIFT score of 0.05 or less and predicted them to be “intolerant.” PolyPhen-2 calculates the probability of causing damage as a score. We selected the mutations that were classified as “possibly damaging,” with a score of 0.15 or higher, and “probably damaging,” with a score of 0.85 or higher. The search for known variants in humans was performed using the Leiden Open Variation Database 3.011,21 and gnomAD browser. 6
Pathological Examination
The collected skeletal muscles were fixed in 10% neutral-buffered formalin. The samples were embedded in paraffin and sectioned according to routine methods, then used for hematoxylin and eosin staining and IHC.
Immunohistochemistry
To clarify the distribution of dystrophin expression and the differences in labeling according to the mutation sites in our cases, IHC was performed using 4 different anti-dystrophin antibodies that recognized different regions of the protein. The following 4 anti-dystrophin antibodies were used as primary antibodies: NCL-DYSB, which recognizes the Rod region located near the N-terminus (mouse monoclonal antibody; clone 34C5; 1:200 dilution; Leica Biosystems, Newcastle, UK); NCL-DYS1, which recognizes the center of the Rod region (mouse monoclonal antibody; clone Dy4/6D3; 1:100 dilution; Leica Biosystems); ab15277, which labels the C-terminus (rabbit polyclonal antibody, 1:500 dilution; Abcam, Waltham, Massachusetts); and MAB1645, with an unknown recognition site (mouse monoclonal antibody; clone 1808; 1:100 dilution; Merck, Darmstadt, Germany). Deparaffinized tissue sections were incubated with 0.3% hydrogen peroxide in methanol at room temperature for 30 minutes to block endogenous peroxidase. For antigen retrieval, specimens were boiled in 1 mM EDTA buffer (pH 8.0; for NCL-DYSB), microwaved in citric acid buffer (pH 6.0) for 15 minutes each (for NCL-DYS1 and ab15277), and autoclaved in an antigen-activation solution (pH 9; NICHIREI BIOSCIENCES, Tokyo, Japan) at 120°C for 20 minutes (for MAB1645). The tissue sections were incubated with a 4% solution of Block Ace Powder (KAC Co., Ltd., Hyogo, Japan) at room temperature for 30 minutes to prevent nonspecific binding. Subsequently, these sections were incubated with the primary antibodies at 4°C overnight. Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgG polymers (Histofine Simple Stain MAX-PO [MULTI]; NICHIREI BIOSCIENCES) were used as the secondary antibodies and applied at room temperature for 30 minutes. Finally, 3,3′-diaminobenzidine (DAB; Wako, Tokyo, Japan) was used as the chromogen and hematoxylin was used for counterstaining.
The IHC results for dystrophin in each case were scored on a 4-point scale. In all 4 antibodies, sarcolemma of all myofibers were strongly positive in normal animals. The 4-point scale was defined as follows: 0 if all sarcolemma are positive, as in normal animals; 1 if >90% of myofibers are positive, locally attenuated; 2 for a mixture of positive and negative fibers, with a frequency of negative fibers between 10% and 50%; and 3 for negative fibers in >50% of cases.
Western Blot
The frozen muscle samples were mixed with a 2% SDS solution and crushed using a Micro smash MS-100R instrument (TOMY, Tokyo, Japan), followed by centrifugation, the addition of benzonase (Merck), and incubation at room temperature for 30 minutes to prepare solubilized samples. The protein concentration in each sample was measured using the DC Protein Assay Kit (Bio-Rad, Hercules, CA), and 10 µg of protein was later applied per SDS-PAGE lane. These samples were diluted in sample buffer (AE-1430 EzApply; ATTO, Tokyo, Japan) and heated to 100°C for 15 minutes, to prepare them for SDS-PAGE.
Proteins (10 µg; 20 µl) were separated in 7.5% or 5% to 20% gradient polyacrylamide gels (ATTO). After separation, the proteins were transferred onto PVDF (polyvinylidene difluoride) membranes (Bio-Rad), which were then blocked with 5% skim milk powder in phosphate-buffered saline containing 0.1% Tween 20. The membranes were incubated with the following anti-dystrophin antibodies at 4°C overnight: MAB1645 (1:500 dilution) and ab15277 (1:1000 dilution). On the following day, the membranes were washed and incubated with the following secondary antibody: HRP-conjugated anti-mouse immunoglobulin (rabbit polyclonal; 1:1000 dilution; Dako) or anti-rabbit immunoglobulin (goat polyclonal; 1:1000 dilution; Dako) for 40 minutes at room temperature. After washing, the bound antibodies were visualized using the Immobilon Western Chemiluminescent HRP Substrate (Merck). Images were captured using an LAS 4000 mini system (GE-HealthCare, Chicago, Illinois).
Results
Dystrophin Gene Sequencing Analysis
The sequence analysis of the dystrophin cDNA revealed the presence of 14 missense mutations and 3 types of shortened transcripts with exon skipping. Two mutations were selected by SIFT 4 and 5 by PolyPhen-2 14 as missense mutations that were suspected to be involved in the pathogenesis of the disease. All detected mutations and their details are summarized in Table 2.
Summary of the detected dystrophin mutations in pigs.
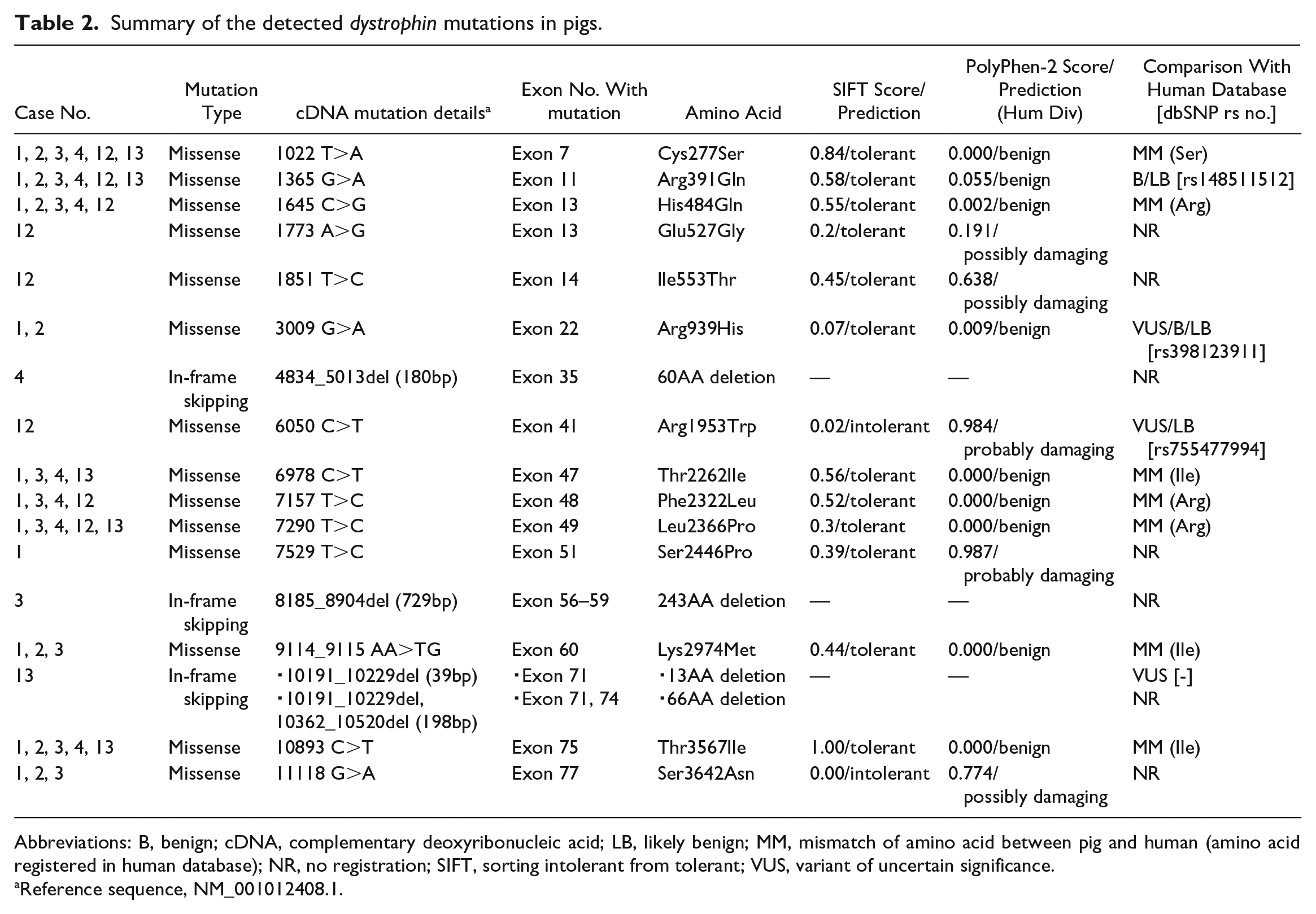
Abbreviations: B, benign; cDNA, complementary deoxyribonucleic acid; LB, likely benign; MM, mismatch of amino acid between pig and human (amino acid registered in human database); NR, no registration; SIFT, sorting intolerant from tolerant; VUS, variant of uncertain significance.
Reference sequence, NM_001012408.1.
The Arg1953Trp mutation, which was observed only in case 12, is listed in the human database as being of uncertain significance or likely benign;6,21 however, it is comparable to a mutation previously detected in a pig pedigree with dystrophinopathy.15,30 Other missense mutations suspected of being involved in pathogenesis were not registered in the human database.6,21 In addition, case 12 had concurrent Glu527Gly and Ile553Thr mutations, which were suspected to be involved in the pathogenesis of the disease by the in silico program. Two suspicious missense mutations, Ser2446Pro and Ser3642Asn, were detected in case 1, whereas Ser3642Asn and transcripts without exon 35 were detected in case 3, concurrently. For all point mutations observed in the 2 female cases, including those that were not suspected to be involved in the pathogenesis, only the mutant allele and no consensus bases were detected in the sequencing data. This indicated that the single nucleotide mutations in the female cases were homozygous.
Regarding the shortened transcripts detected in this study, short- and normal-sized transcripts were also obtained for all regions, and short transcripts were generated using in-frame exon skipping (Table 2). A shortened transcript without the full exon 9, which was identified in all cases, was registered as one of the predicted isoforms in pigs (XM_021079553.1 and XM_021079555.1). These transcripts were also obtained in the normal pigs. The 3 transcripts listed in Table 2 were not registered as pig isoforms. In case 13, 2 shortened transcripts were detected: one lacking all bases encoding the 13 amino acids of exon 71, and the other lacking all bases that corresponded to the 66 amino acids of exons 71 and 74, respectively. Skipping of exon 71 has been reported as a variant of unknown significance in a human patient with BMD; however, no transcript with simultaneous skipping of exons 71 and 74 has been observed in such patients.21,34 Other mutations accompanying shortened transcripts identified in this study were not previously registered in humans.
Histopathology of Porcine Muscle Tissues
Fatty replacement was observed among the myofibers in all cases (Fig. 2a), and numerous myofibers with increased eosinophilia and a rounded shape (opaque fibers) were detected. The myofibers were observed diffusely and were enlarged up to approximately twice the diameter of the normal, angular muscle fibers with normal staining (Fig. 2b). Macrophages infiltrated necrotic myofibers (Fig. 2b) and there were also areas of coagulation necrosis without macrophages. Regenerative changes, such as fiber splitting (Fig. 2c), nucleus centralization, and regenerative myofibers with small diameters and basophilia (Fig. 2d) were also observed. Similar lesions were detected in the longitudinal sections of the muscle, with disorder of the arrangement of hypertrophic muscle fibers.
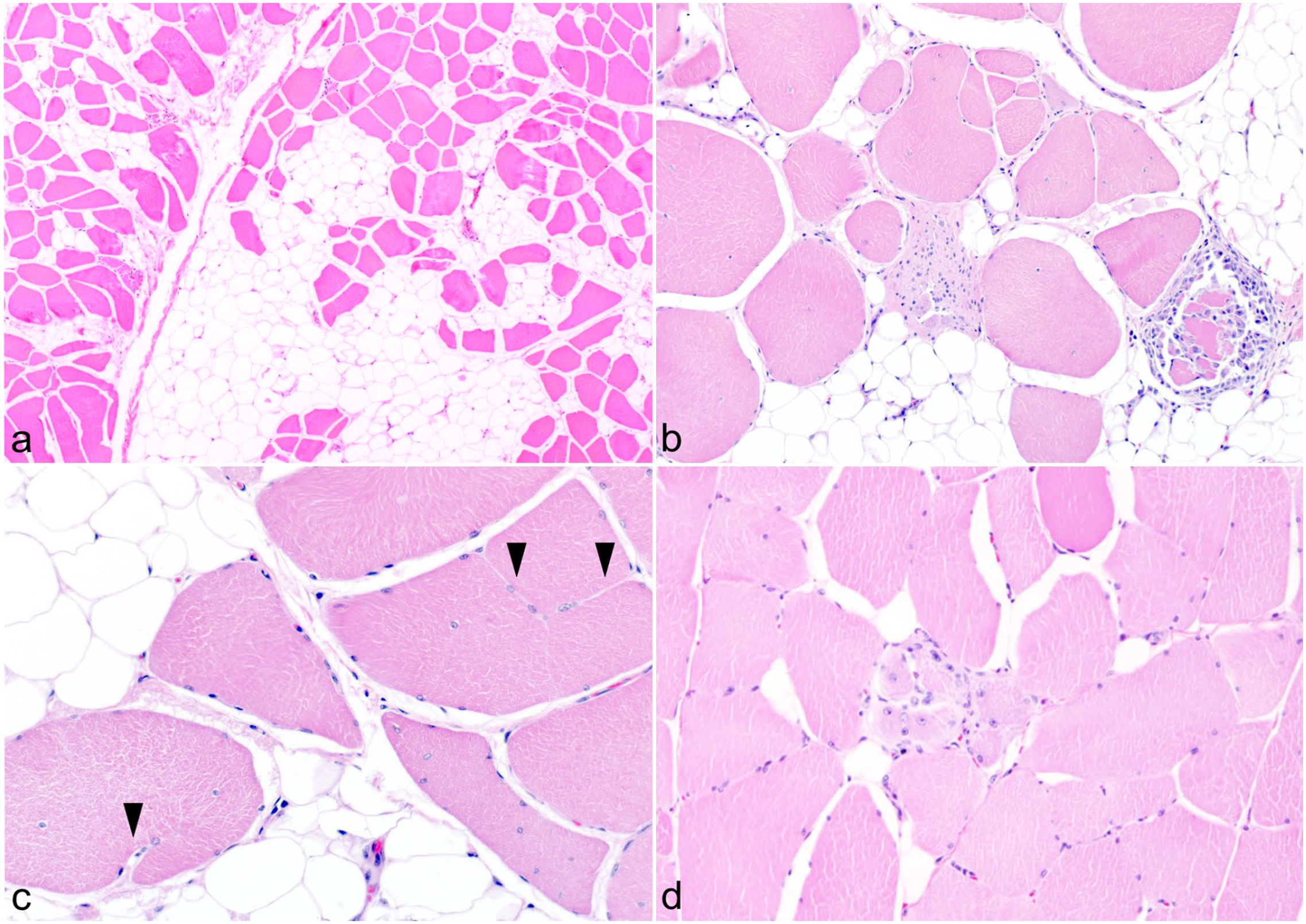
Dystrophinopathy in the gracilis muscle of a pig. Hematoxylin and eosin. (a) Fatty replacement among myofibrils, case 3. (b) Unequal myofibril diameters and necrotic myofibers infiltrated by macrophages, case 1. (c) Nucleus centralization and fiber splitting (arrowhead), case 1. (d) Regenerative myofibers that are small, basophilic, and have centralized nuclei, case 12.
There was no clear difference in the degree of lesions between males and females.
Immunohistochemistry of Porcine Muscle Tissues
The IHC results showed that 10 out of the 25 cases had attenuated dystrophin labeling with all the antibodies, followed by 4 cases with MAB1645 and ab15277, and 3 cases with MAB1645 only. The other 8 cases had attenuated labeling for 1 to 3 antibodies. Details of the scores obtained for each case are provided in Table 3.
Summary of immunohistochemistry results.
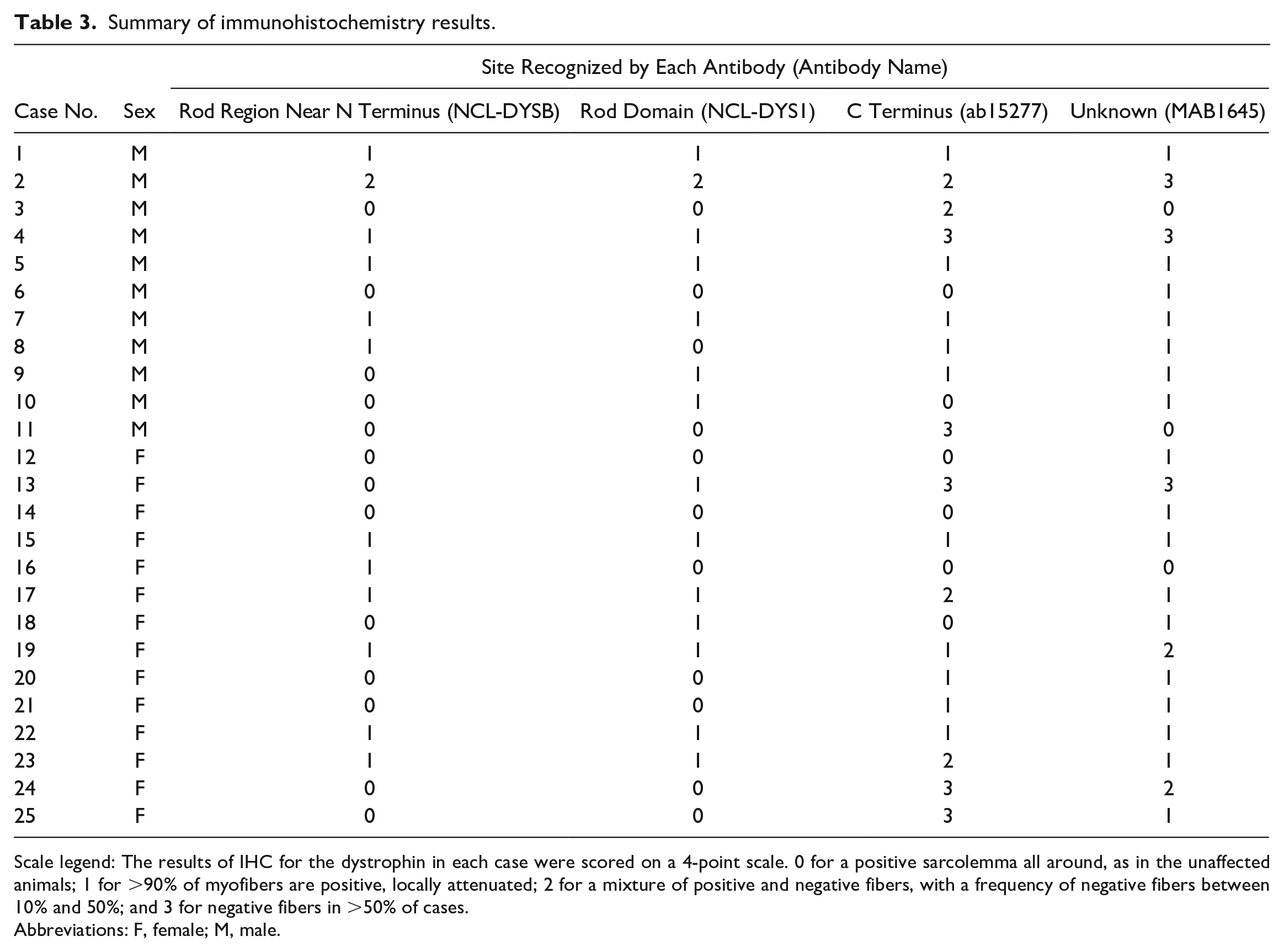
Scale legend: The results of IHC for the dystrophin in each case were scored on a 4-point scale. 0 for a positive sarcolemma all around, as in the unaffected animals; 1 for >90% of myofibers are positive, locally attenuated; 2 for a mixture of positive and negative fibers, with a frequency of negative fibers between 10% and 50%; and 3 for negative fibers in >50% of cases.
Abbreviations: F, female; M, male.
The comparison of the results obtained for each antibody revealed that the majority of cases showed attenuation when using MAB1645, with an unknown recognition site. Immunolabeling with the 2 antibodies that recognize the Rod region tended to reveal a relatively preserved expression, whereas labeling with the C-terminus-recognizing antibody showed a clear decrease in expression in many cases. An evaluation score of “3” was obtained exclusively for ab15277 and MAB1645.
The IHC results revealed no apparent sex differences related to the trend and degree of labeling attenuation according to each antibody.
Among the cases in which a genetic analysis could be performed, cases 1 and 2 showed decreased labeling with all antibodies (Fig. 3a–d), and the regions and labeling patterns of attenuation in each case were similar among the antibodies. Case 3 exhibited downregulation only for the C-terminal-labeling antibodies (Fig. 3e–h). Cases 4 and 13 showed normal or mostly positive myofibers for the antibodies recognizing the Rod region although the labeling intensity was uneven; whereas, the C-terminus-recognizing antibodies showed a marked decrease in labeling (Fig. 3i–l). Case 12 showed decreased labeling only for antibodies with unknown recognition sites (Fig. 3m–p).
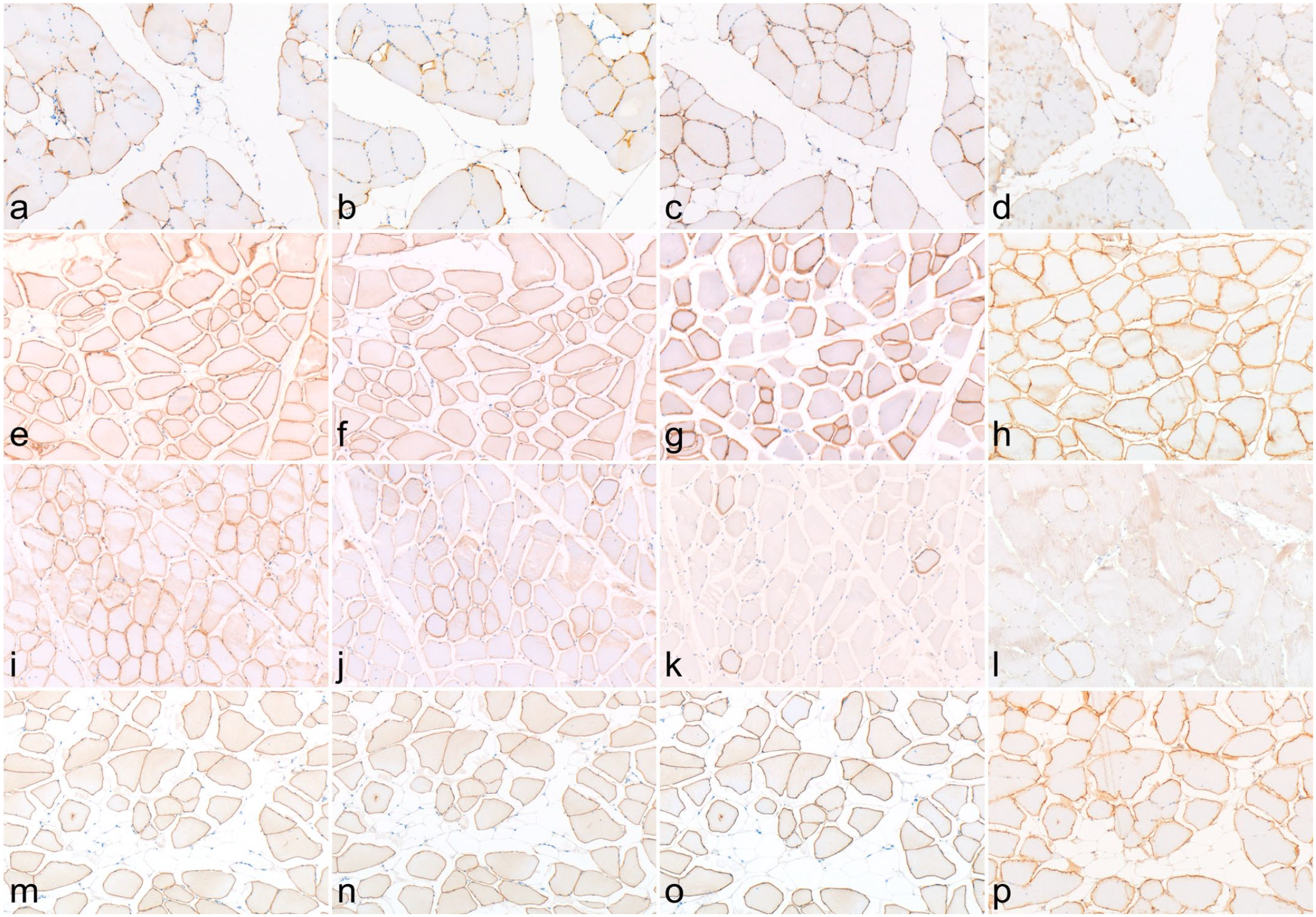
Dystrophinopathy in a skeletal muscle of a pig. Immunohistochemistry for dystrophin. (a, e, i, and m) NCL-DYSB antibody. (b, f, j, and n) NCL-DYS1 antibody. (c, g, k, and o) ab15277 antibody. (d, h, l, and p) MAB1645 antibody. (a–d) case 2. (e–h) case 3. (i–l) case 4. (m–p) case 12.
Western Blot
Western blotting was performed to determine the extent of dystrophin expression and its size in 6 of our cases. Using MAB1645 (with an unknown recognition site) as the primary antibody, we observed decreased expression of a product that migrated at a normal size of 425 kDa in all affected pigs (Fig. 4a). In cases 1 to 3, 12, and 13, a band was observed at around 31 kDa (or at less than 31 kDa in the 5%–20% gradient gel). In case 4, a band located at 55 to 71 kDa was observed.
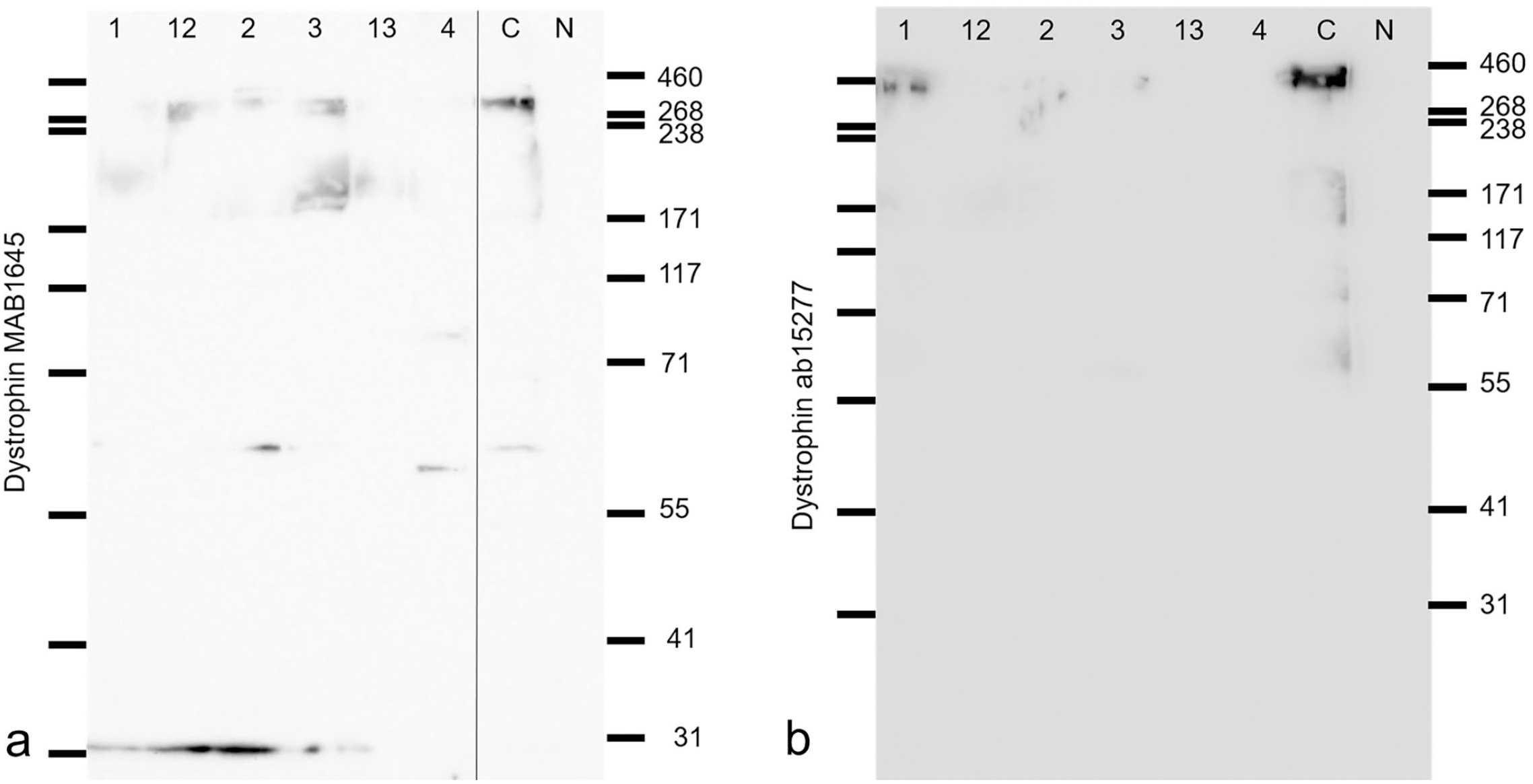
Dystrophinopathy in a skeletal muscle of a pig. Western blot for dystrophin; cases 1 to 4, 12, and 13. (a) MAB1645 antibody (7.5% polyacrylamide gel). (b) ab15277 antibody (5%–20% gradient polyacrylamide gel).
Using the antibody that recognizes the C-terminus of dystrophin, we observed 2 bands at approximately 425 kDa in the control, of which the upper band was thought to be at 425 kDa. In all cases, the expression level of the normal-sized dystrophin was markedly decreased. Two bands were obtained in case 1, as in the control, and an indistinct band was obtained in cases 2, 3, and 12 (Fig. 4b). Finally, in cases 4 and 13, the expression of dystrophin was markedly decreased and no bands could be identified.
Discussion
In this study, we identified 14 missense mutations and 3 types of exon skipping, resulting in shortened transcripts in the cDNA of the porcine dystrophin gene. Pathogenetic involvement was suspected in 5 of the missense mutations and in the 3 patterns of shortened transcripts caused by exon skipping. One missense mutation had been previously reported,15,30 whereas the remaining mutations identified here had not been previously documented in pigs; thus, the diversity of the dystrophin gene mutations in pig populations was confirmed.
One missense mutation, Arg1953Trp, which was detected in case 12, had been reported previously in the United States as causing a condition similar to swine stress syndrome.15,30 The existence of the same missense mutations in both the United States and Japan suggests that these mutations are prevalent across diverse pig populations. Moreover, the results of the in silico analyses suggest that these mutations are involved in the pathogenesis of the disease; however, some animals carried more than one suspected mutation. Therefore, the evaluation of additional cases is needed to determine the importance of these mutations.
Normal and shortened transcripts were confirmed in cases 3, 4, and 13. Because cases 3 and 4 were male and, thus, had only one X chromosome, splicing abnormalities were suspected. In human dystrophinopathies, most cases are caused by deletions or duplications, and splicing mutations are rare.1,18,21,38 In contrast, a pseudoexon insertion was also considered to be a splicing abnormality in a previously reported porcine case. 3 Therefore, here we showed that splicing abnormalities may be a major class of mutation in porcine dystrophinopathy, and confirmed the uniqueness of dystrophin gene mutations in pig populations.
In humans, most dystrophin splicing mutations are caused by alterations in the 5’ and 3’ splice sites in introns, 5 and few cases are caused by mutations in exons that disrupt the cis-acting elements of splicing.9,12,32,36 In this study, in the pigs for which short transcripts were obtained, no mutations in the normal-length transcripts corresponding to the region of exon skipping were detected. Therefore, this type of mutation was probably caused by alterations in the 5’ and 3’ splice sites of introns; thus, a search for introns is necessary to confirm this mutation.
Exon 71 skipping in a human with BMD was detected through droplet digital PCR and was accompanied by a normal-sized transcript with no apparent mutations at the RNA level, suggesting a mutation in the intron region. 34 The patient presented with mild symptoms, and the significance of the skipping is unknown, 34 but this case was thought to be similar to the case reported in this study. As the human case showed a low percentage of shortened transcripts (16%), 34 we considered it necessary to closely examine the relationship between the percentage of shortened transcripts and the disease pathogenesis.
The short transcripts produced by skipping exons 35, 71, or 74 in the present cases were registered previously as human pathogenic mutations that are caused by the deletion of introns 34–35,21 –23,42 70–71,21,40 or 73–7421,41 across the skipped exon. In the present cases, there was no deletion of the exon in the genome; therefore, the actual mutation was different. However, based on the registered human cases, the shortened transcripts are thought to affect the structure and function of the protein, suggesting that they are involved in the pathogenesis of the disease. Although the regulatory mechanism governing the transcription ratio of abnormal and normal transcripts remains unclear, the expression of residual wild-type dystrophin mRNA in such cases is likely to result in a phenotype consistent with BMD.
All cases were considered to have a BMD-like phenotype, considering that the IHC results showed a decrease in positive regions, albeit with retention of protein expression. In this study, differences in the degree of labeling attenuation were observed according to the primary antibody used for IHC; in turn, differences in IHC results have also been reported in human BMD cases when multiple antibodies are used. The use of multiple anti-dystrophin antibodies is recommended for the evaluation of dystrophin in suspected BMD-like myopathy cases.
In this study, no clear relationship was found between each of the detected mutations and IHC results for each primary antibody. This was thought to be because the mutations contributing to the pathogenesis did not match the recognition region of the primary antibodies used in this study (Supplemental Fig. S1). Comparing the IHC results, the majority of the examined cases exhibited decreased labeling with the C-terminus-recognizing antibodies. This suggests a mutational effect because the C-terminus is located downstream of all of the detected mutations. The NCL-DYSB and NCL-DYS1 antibodies recognize exons 10 to 13 and 26 to 30, respectively, and had a score of ≤2 in each case. This suggested that these regions are located upstream of most of the detected mutations and were less affected.
Western blotting revealed a reduction in the amount of normal-sized dystrophin, supporting the IHC results. Although multiple exon skipping was observed in the sequencing analysis in this study, the molecular weights of the proteins detected by western blot were smaller than expected, considering the number of amino acids that were expected to be deficient due to exon skipping in each case. In humans with dystrophinopathies, western blot results may show a multiple-band pattern. 28 It is possible that the small bands obtained in this study were also degraded in vivo because of the instability of the translated protein. In addition, because PCR and sequencing were performed by subdividing the full length of the dystrophin cDNA, it was possible that there were deletions across each amplified region.
Pathologically, fatty replacement, degeneration, and necrosis of muscle fibers were commonly observed in porcine dystrophinopathy. In humans, pseudohypertrophy is reportedly more pronounced in BMD, 20 thus, the pronounced fatty replacement observed in pigs was thought to be consistent with the pathogenesis of BMD in humans. Because we were unable to evaluate the entire body, investigating the extent and distribution of the muscular lesions in the present cases was difficult. However, some pigs showed fatty replacement in skeletal muscles throughout the body grossly, suggesting that lesions were systemic.
Here, both sexes showed decreased dystrophin protein expression by IHC and Western blot, to varying degrees. In humans, muscle lesions are present in female carriers of BMD and carriers of DMD.8,13 The manifestations of BMD in female carriers are diverse, and the timing of development of clinical signs and the extent of lesions also vary. 8 Although mutations could be analyzed in only 6 cases, the variety of mutations detected and the fact that the IHC results and WB results did not differ between males and females were considered to be consistent with the findings of the pathological evaluation.
In addition, more than half of the cases examined in this study were females, which deviated from the fact that 2.5% to 17% of female carriers of BMD/DMD are symptomatic in humans.8,16,31 The reason why more female cases were detected in this study remains unclear because of the limited clinical information and number of cases. Meat inspection for pigs is typically performed at a young age; thus, the stage of the disease we searched for in pig cases may correspond to the stage at which human patients are asymptomatic, but the damage to muscular tissue is grossly visible. In addition, the point mutations in female cases were homozygous, and it was expected that those mutations would be retained in parent pigs of both sexes. This may be related to the reason for the high proportion of female cases in this study.
This study revealed the diversity of mutations in the dystrophin gene in porcine dystrophinopathies. The mutations detected here were missense mutations and splicing abnormalities caused by exon skipping, and the phenotype was similar to the human BMD. There appeared to be minimal clinical effects in these pigs at the age of 6 months, as they were not culled during the fattening process. However, they exhibited pale discoloration at a degree that could be detected grossly, which is considered to be a serious economic problem. The examination of the effects of each mutation on motor function and meat quality, respectively, will require the accumulation of additional cases.
Supplemental Material
sj-pdf-1-vet-10.1177_03009858231214028 – Supplemental material for Diversity of mutations in the dystrophin gene and details of muscular lesions in porcine dystrophinopathies
Supplemental material, sj-pdf-1-vet-10.1177_03009858231214028 for Diversity of mutations in the dystrophin gene and details of muscular lesions in porcine dystrophinopathies by Yumiko Kamiya, Naoyuki Aihara, Takanori Shiga, Noriyuki Horiuchi and Junichi Kamiie in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Center for Diversity, Equity & Inclusion, Azabu University, and The Ito Foundation research grant project.
Supplemental Material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.