
Abstract
Fluorescein-derived fluorochromes and anionic dyes such as Fluoro-Jade (FJ) stains have been introduced to facilitate recognition of dying neurons in tissue sections. However, the definition of what is really detected by FJ-based stains and its sensitivity in the detection of neuronal cell death is unclear. In our work, we evaluated the outcome of FJ-C staining in mouse brains from 4 different well-characterized models of neurodegeneration. Neuronal degeneration and loss were highlighted with high sensitivity by FJ-C stain in mice with dysfunctional γ-secretase in the glutamatergic neurons and in mice affected by acute cerebral ischemia. Histopathologically, acute eosinophilic necrosis or “red dead” neurons were associated with FJ-C staining in both settings. Conversely, in mice affected by chronic cerebral microinfarcts due to tumor lysis syndrome as well as in a model of mitochondrial encephalopathy, FJ-C staining failed to detect neuronal death. Histopathologically, these models were characterized by extensive neuronal vacuolation associated with fading neurons (“ghost cells”). Therefore, contrary to the widespread belief that FJ-C stain has high affinity for all degenerating neurons regardless of the underlying cell death mechanism, we observed restricted sensitivity of the technique to specific conditions of neuronal cell death. As such, complementary techniques are essential to evaluate the presence of neurodegeneration in the absence of a positive FJ-C signal.
Detecting neuronal degeneration is a priority to understand the pathogenesis of neurological diseases and to evaluate the effects of novel drugs on the central nervous system. Historically, pathologists relied heavily on conventional detection methods such as hematoxylin and eosin (HE) and Nissl stains to detect degenerating neurons based on their morphology or changes in staining intensity. 15,27 These methods come with technical challenges and disadvantages including limited sensitivity and ambiguous interpretation of the morphological findings. Thus, the need of a reliable, reproducible, and highly sensitive histochemical marker of neuronal degeneration became apparent. 27
Fluorescein-derived fluorochromes and anionic dyes such as Fluoro-Jade (FJ) were introduced in the late 1990s to facilitate the recognition of neuronal degeneration resulting from exposure to a variety of neurotoxic insults. 27,29 Since the discovery of FJ, the analogs Fluoro-Jade B (FJ-B) and C (FJ-C) were developed with an increased affinity and resolution for all the different compartments of degenerating neurons (cell body, dendrites, axons, and terminals). 28,30 FJ dyes were designed specifically to stain only degenerating neurons regardless of the types of neurotoxic insults or trauma, thus reducing the false positive results to a negligible minimum. 25 However, information on the biochemical principles that allow these dyes to selectively label dying neurons remains largely unknown. 22
A correct understanding of the usefulness and limitations of FJ dyes in association with morphologic neuronal changes is pivotal to adequately diagnose neuronal loss in different mouse brain conditions. To this aim, here we report the results of a retrospective study investigating the sensitivity of FJ-C in paraffin-embedded brain samples from different experimental mouse models characterized by neuronal degeneration and death or loss. While this stain has been deemed extremely sensitive for neuronal degeneration regardless of the underlying cause, 4,18,30 here we show that its sensitivity is not universal but rather dependent on the specific mechanisms of neurodegeneration.
Materials and Methods
For this study, paraffin-embedded brain samples from different preclinical mouse models were used. These included models of (1) dysfunctional γ-secretase in glutamatergic neurons (experimental model 1; B6.129P2(Cg)-Tg(Camk2a-cre)T29-1Stl Aph1atm1Bdes Aph1btm1Bdes Aph1ctm1Bdes/J mice), (2) transient cerebral ischemia (experimental model 2; C57BL/6J mice), (3) cerebral microinfarcts secondary to tumor lysis syndrome (experimental model 3; NOD.Cg-Prkdcscid Il2rgtm1Wjl/J mice), and (4) mitochondrial encephalopathy (experimental model 4; B6.129P2(Cg)-Parltm1.1Bdes/Ieg mice). The animals were utilized according to the experimental contexts as described in 1,13,16,32 and also reported in Table 1.
Samples were selected based on the reported evidence of brain lesions characterized by neuronal degeneration and neuronal death or loss as previously demonstrated by detailed neuropathological analysis and further confirmed by immunohistochemistry (IHC) for glial and/or neuronal markers. The selection of brain areas for FJ-C staining in the different models reflected the severity of histopathologic changes. Mice with dysfunctional γ-secretase in glutamatergic neurons and mice with mitochondrial encephalopathy were analyzed at serial time points ranging from the initial phase to the late stage of the disease. A single time point was considered for mice with transient cerebral ischemia and mice with cerebral microinfarcts secondary to tumor lysis syndrome (Table 1).
Main Features and Highlights of the Investigations Conducted in Experimental Mouse Models
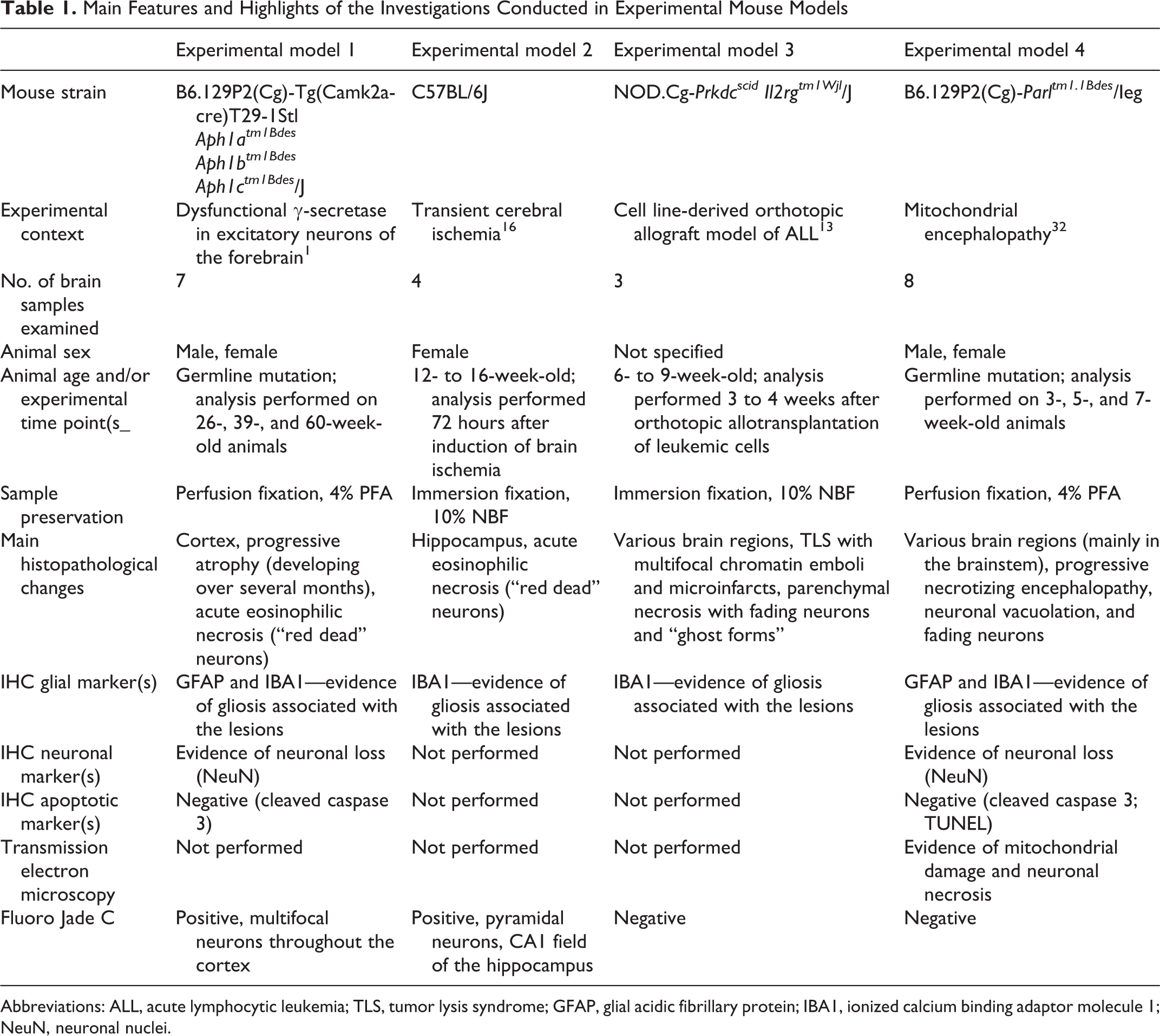
Abbreviations: ALL, acute lymphocytic leukemia; TLS, tumor lysis syndrome; GFAP, glial acidic fibrillary protein; IBA1, ionized calcium binding adaptor molecule 1; NeuN, neuronal nuclei.
Table 2 reports the details concerning primary antibodies and procedures used for immunohistochemistry and immunofluorescence. The TUNEL assay was performed using the In Situ Cell Death Detection Kit (Millipore-Sigma, Catalog #11684795910) according to the manufacturer’s instructions. The protocol for transmission electron microscopy is described elsewhere. 32 FJ-C staining was performed on 10-μm-thick sections according to manufacturer’s recommendation. (https://www.emdmillipore.com/US/en/product/Fluoro-Jade-C, MM_NF-AG325). DAPI nuclear counterstaining was included in the protocol (Sigma-Aldrich D9542). Paraffin sections of the hippocampus with acute neuronal death from a porcine model of cardiac arrest and cardiopulmonary resuscitation were included as positive control. 21 Paraffin sections of brains from wild-type mouse littermates (for the 2 genetic models considered in this study) or clinically normal and experimentally naïve C57BL/6J were included as negative controls.
Details Concerning Reagents and Procedures Used for Immunohistochemistry/Immunofluorescence.
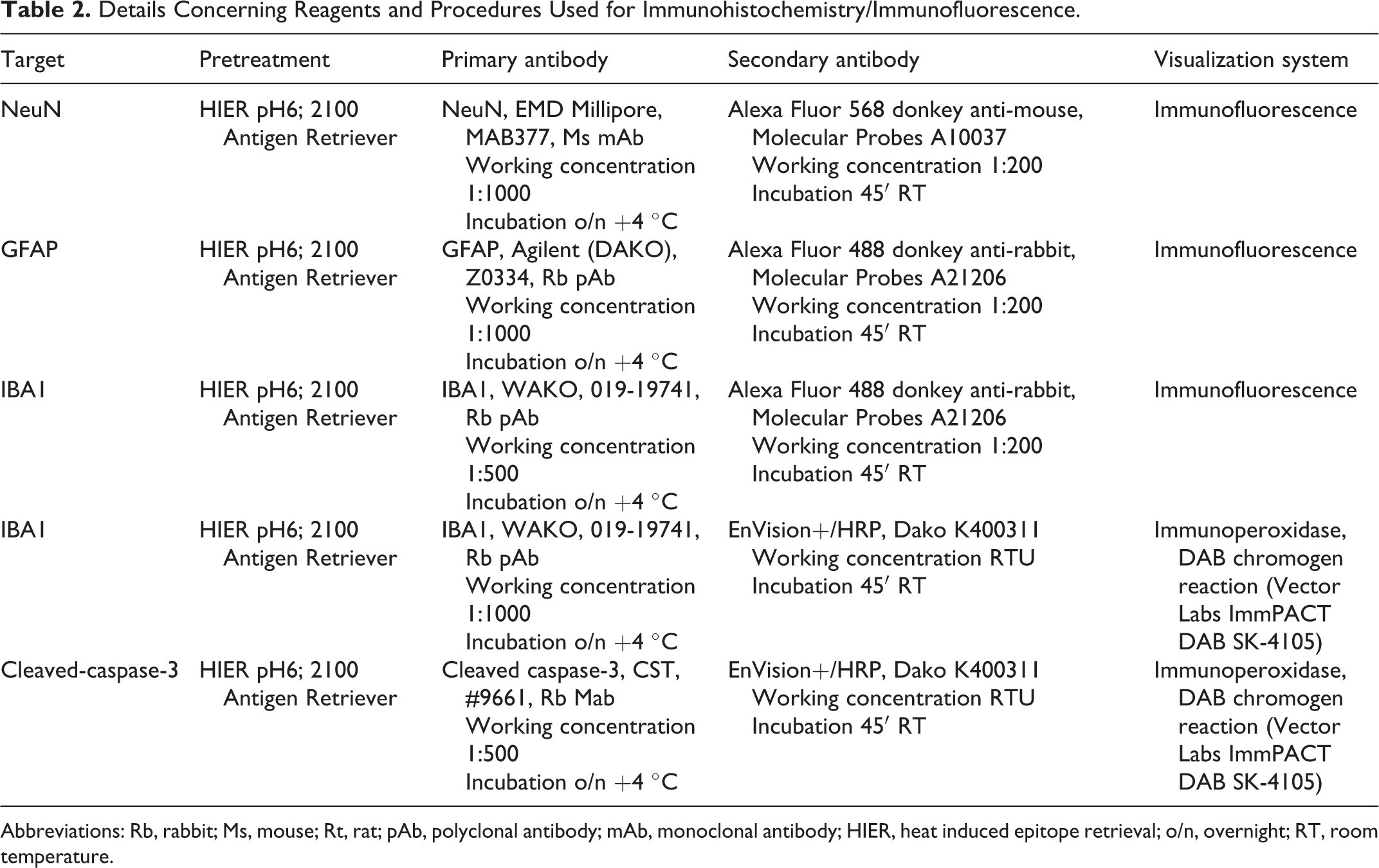
Abbreviations: Rb, rabbit; Ms, mouse; Rt, rat; pAb, polyclonal antibody; mAb, monoclonal antibody; HIER, heat induced epitope retrieval; o/n, overnight; RT, room temperature.
Results
For each experimental model considered in this study, the salient neuropathological changes including the results of FJ-C stain and any additional IHC investigation are summarized in Table 1. Compared to HE, neuronal degeneration and loss were highlighted and enhanced by FJ-C stain in mice with dysfunctional γ-secretase in the glutamatergic neurons and in mice affected by acute cerebral ischemia (Figs. 1, 2). Histopathologically, both these models were characterized by acute eosinophilic necrosis (“red dead” neurons; Fig. 3) with gliosis (Fig. 4). While FJ-C signal was mainly observed within the perikarya of the neurons, individual neurites were also highlighted by the stain in some instances. In mice with dysfunctional γ-secretase, progressive neuronal loss was also confirmed by means of NeuN IHC (Suppl. Fig. S1). In the same model, evaluation of serial brain sections stained with HE and FJ-C demonstrated significantly more FJ-C-positive neurons compared to the “red dead” neurons visible with HE stain (Suppl. Fig. S2).
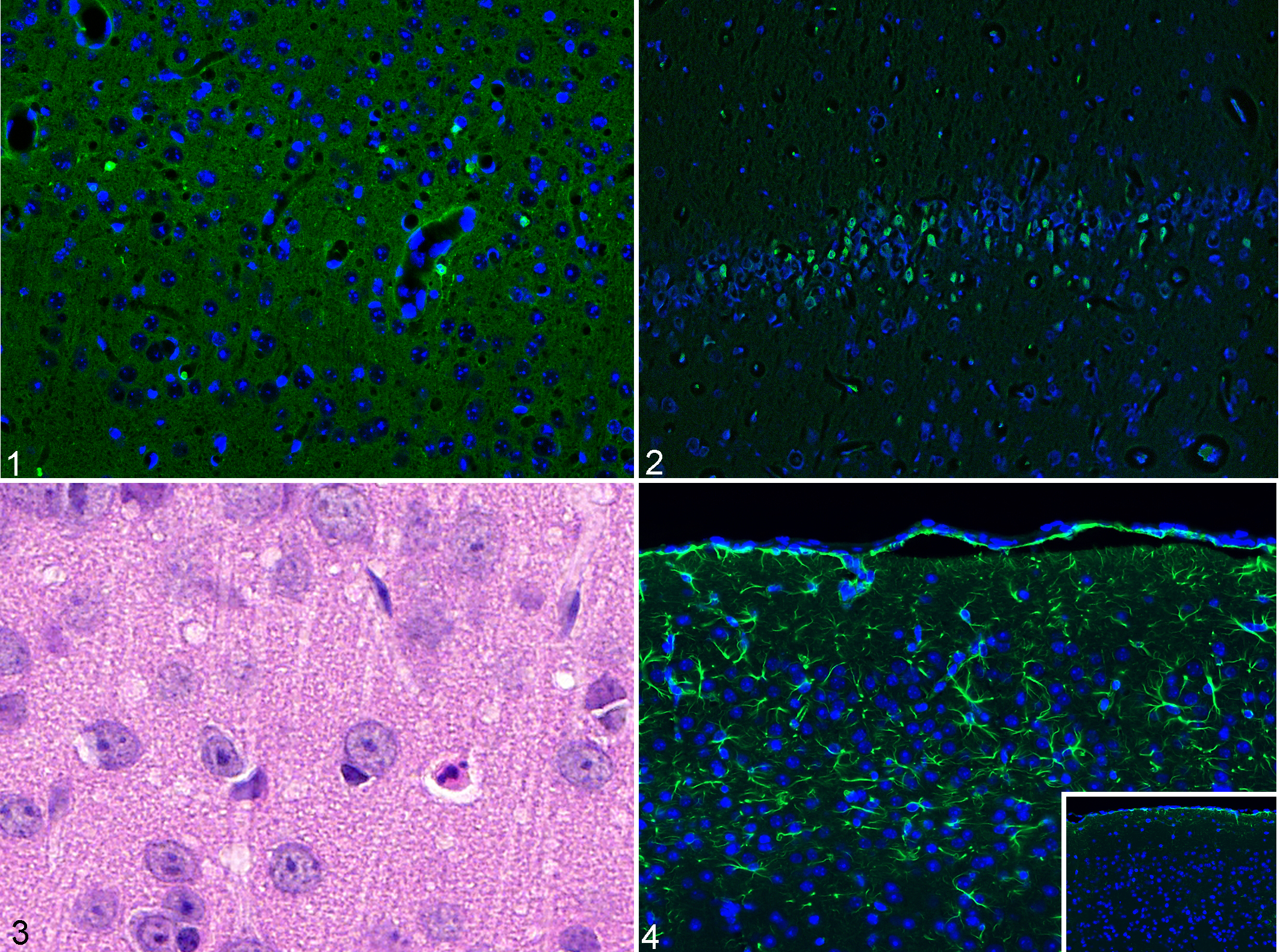
Mouse model of dysfunctional γ-secretase in excitatory neurons of the forebrain (Figs. 1, 3, and 4) and mouse model of transient cerebral ischemia (Fig. 2); brain, mouse.
Conversely, in mice affected by chronic cerebral microinfarcts due to tumor lysis syndrome as well as in the model of mitochondrial encephalopathy, degenerated neurons were not stained by FJ-C (Figs. 5, 6) despite the presence of severe parenchymal necrosis with fading neurons (“ghost cells”) as well as extensive neuronal vacuolation (especially in the case of mitochondrial encephalopathy; Figs. 7, 8). Gliosis and neuronal loss were confirmed by IHC (Suppl. Fig. S3) and, in mice with mitochondrial encephalopathy, by electron microscopy (Fig. 9). 32 No NeuN staining was performed in mice with cerebral microinfarcts secondary to tumor lysis syndrome.
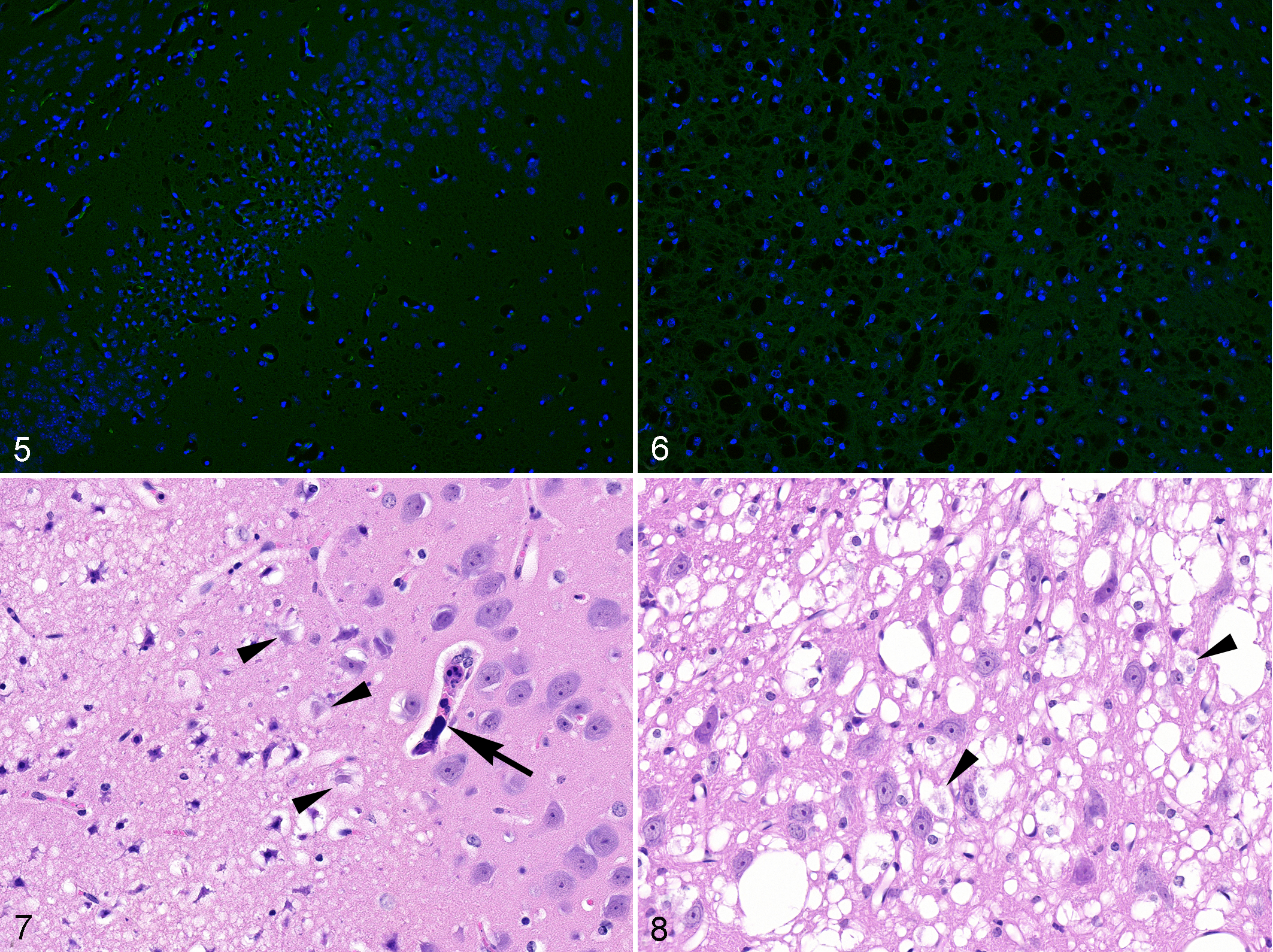
Mouse model of tumor lysis syndrome (Figs. 5, 7) and mouse model of mitochondrial encephalopathy (Figs. 6, 8); brain, mouse.
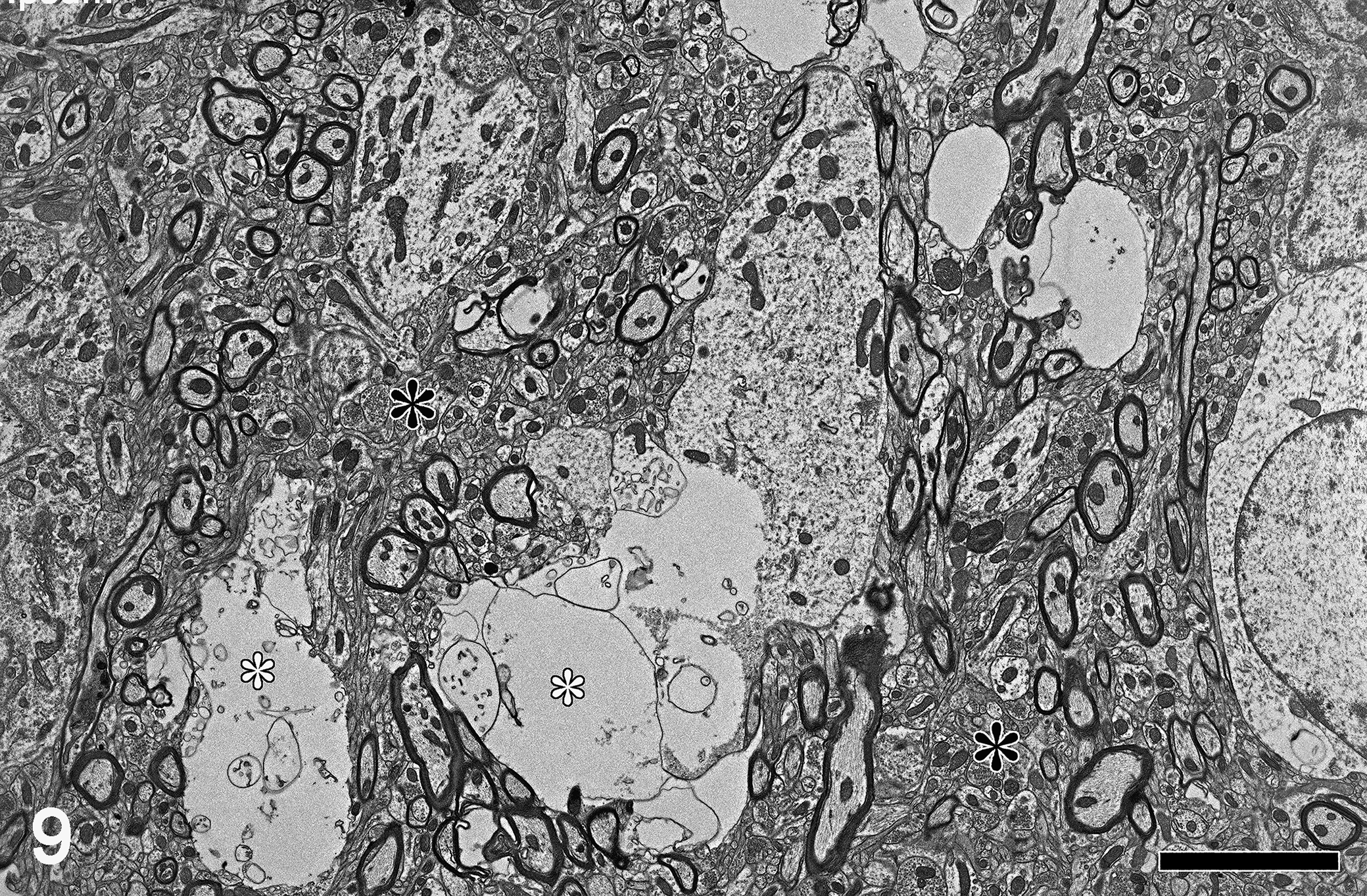
Mouse model of mitochondrial encephalopathy; brain (thalamus), mouse. Degenerating neurons display partial cytoplasmic rarefaction with loss of intracellular organelles and accumulation of abundant electron-lucent material (white asterisks) compartmentalized by distended membranous profiles (neuronal vacuolization). Neuropil surrounding affected neurons contains an intricate mesh of dendritic projection and myelinated axons (black asterisk). Transmission electron microscopy. Scale bar: 5 μm.
In mice with dysfunctional γ-secretase in the glutamatergic neurons and in mice with mitochondrial encephalopathy, there was no evidence of apoptosis as assessed by the TUNEL assay, or by cleaved-caspase 3 IHC performed at different time points.
Discussion
Neuronal loss represents the ultimate endpoint of neurological diseases associated with neurodegeneration. Upon examination of HE-stained sections, the identification of degenerating or dying neurons at acute stages (sometimes referred to as “red dead” neurons) is based on the recognition of morphologic features that include hypereosinophilic cytoplasm, cellular shrinkage, and nuclear pyknosis and fragmentation. 8 In addition, the finding of fading cells with pale cytoplasm (so-called “ghost cells”), and the presence of enlarged vacuolated neurons in cases of spongiform encephalopathies and storage diseases, are all morphologies indicative of progressive neuronal degeneration and death.
However, the identification of neuronal degeneration on HE-stained slides provides suboptimal sensitivity. Therefore, selective and unequivocal markers have the potential to improve the accuracy of identifying neuronal necrosis. 4 The fluorescent dye FJ was introduced in 1997 to aid in the detection and localization of toxicant-induced neuronal degeneration. 27 Numerous neuropathological alterations have been demonstrated by using FJ since its discovery. The demonstration of neuronal degeneration with FJ staining has been documented in rodents following exposure to physical trauma, 2,10,36 or to toxic compounds including kainic and domoic acids, the mitochondrial complex II inhibitor 3-nitropropiooinic acid (3-NPA), ibogaine, ketamine, or abuse substances such as phencyclidine, dizocilpine, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. 17,23,27,30 FJ dyes have been also employed for the identification of injured neurons in diverse mouse model of neurodegeneration including epilepsy, 34 ALS, 35 and Parkinson’s disease. 3
In order to improve the strength and long-term stability of fluorescent dyes that bind preferentially to damaged neurons, the next generation of anionic fluorescein derivatives—namely FJ-B 28 and FJ-C 30 —were created. Structurally, all 3 FJ dyes are related. The ability of FJ to stain the entire degenerating neuron, including cell body, dendrites, axon, and axon terminals was maintained in subsequent dyes, in addition to improved resolution and contrast. 22 In particular, the qualitative difference in the staining characteristics of FJ-C translates to a stain of maximal contrast and higher affinity for degenerating or dying neurons.
Similarly to the first generation of FJ, these new dyes have also been universally considered to have high affinity for degenerating or dying neurons, regardless of the specific underlying insult or cell death mechanism. 5,11,24,26,30 Despite the presumed ability of FJ-C to label a broad spectrum of injured and degenerating neuronal cells including mature and immature neurons, neuronal stem/precursor cells and neuroepithelial cells, 12 our work demonstrates that the dye sensitivity is restricted to specific conditions of neuronal cell death. Notably, clear FJ-C positivity was observed in the presence of acute neuronal necrosis (“red dead” neurons) as reported in mice with dysfunctional γ-secretase in the glutamatergic neurons and in mice affected by acute cerebral ischemia (experimental models 1 and 2). Conversely, in mice affected by cerebral microinfarcts (resulting from tumor lysis syndrome) or by mitochondrial encephalopathy (experimental models 3 and 4), fading neurons, and/or neurons exhibiting vacuolar degeneration appeared consistently negative for FJ-C.
The mechanism by which the FJ dyes bind to degenerating or dying neurons is not fully understood. Biochemically, FJ is considered an analogue of eosin. Both are anionic dyes staining acidophilic structures that have a net positive charge. 14 Therefore, considering the characteristic eosinophilic tinctorial properties of acute neuronal necrosis (“red dead” neurons), it can be speculated that the binding affinity of FJ-C is restricted to those conditions characterized by accumulation of positively charged molecules in degenerating or dying neurons. In that context, electrostatic attraction to polyamines produced during tissue degradation (ie, cadaverine, spermidine, and putrescine) has been most recently proposed as a possible biochemical explanation. 25 Further supporting this view, FJ dyes are known to label a variety of acidophilic (eosinophilic) tissue structures including amyloid plaques, 9 Rosenthal fibers, 31,33 gemistocytes, 6,30 apoptotic cells, 12 and red blood cells. 30
Interestingly, we also confirmed that the identification of degenerating or dying neurons was enhanced using FJ-C in comparison to routine evaluation using HE stain. As shown in experimental model 1 (dysfunctional γ-secretase in glutamatergic neurons), only scattered degenerating neurons were noted histologically as opposed to the greater degree of neuronal degeneration apparent upon FJ-C stain.
Importantly, our study clearly indicates that distinct forms of neuronal degeneration and death, such as the case of mitochondrial encephalopathy, cannot be identified by FJ-C regardless of the early or late time point analyzed. However, in specific settings, such as brain injury resulting from altered perfusion, timing and stage of neuronal damage might play an important role in determining FJ-C labeling. In mice with acute cerebral ischemia, FJ-C positivity has been associated with “red dead” neurons, which are considered a manifestation of acute neuronal injury. Contrarily, FJ-C labeling was negative in brains of mice with tumor lysis syndrome and cerebral microinfarcts, which are characterized by fading neurons and “ghost forms,” a morphological manifestation of long-term neuronal damage. Taken together, these findings might suggest the inability of FJ-C to recognize acute forms of neuronal degeneration at later time points. Unfortunately, because of the observational nature of our study, these considerations remain speculative and properly planned time course experiments are necessary to address the persistence of FJ-C labeling at different time points after acute neuronal injury.
Last, differences in the staining ability of FJ-C may rely on the involvement of specific cell death pathways and associated molecules for neuronal conditions. Over the last decades, it became apparent that multiple regulated cell death (RCD) mechanisms are involved in the maturation, differentiation, and plasticity of neurons, 7 as well as in neuronal degeneration and loss in several pathologic conditions of the nervous system. In the Parl-deficient mouse model (experimental model 4), characterized by severe mitochondrial respiratory chain dysfunction due to coenzyme Q10 and complex III deficiency, the negative results of TUNEL and cleaved-caspase 3 in conjunction with evidence of neuronal vacuolization, swelling, and breakdown indicated necrosis as the underlying mechanism of cell death. 32 Although the totality of microscopic and ultrastructural changes were indicative of severe necrosis, the results of FJ-C staining were consistently negative. Conversely, FJ-C has been reported to be positive in a toxicological model of mitochondrial neurodegeneration induced by the complex II inhibitor NPA. 30 Therefore, FJ-C is not sensitive in recognizing all types of neuronal cell death caused by mitochondrial dysfunction. Moreover, in genetically engineered mice with γ-secretase dysfunction in glutamatergic neurons of the forebrain (experimental model 1), cortical neurodegeneration was corroborated by the identification of FJ-C staining. While the underlying mechanism of the γ-secretase model is not completely understood, impaired proteolysis of long-lived proteins together with alterations in other markers of autophagy have been proposed as possible causes of neuronal injury. 19,20
While all the mouse brain conditions discussed herein exhibited clear microscopic lesions compatible with neuronal degeneration, FJ-C staining was only evident in cases with acute eosinophilic neuronal degeneration or death. In addition, the variable outcome of the FJ-C staining in the presence of neuronal degeneration supports the hypothesis that fluorochrome-based anionic dyes may label substances deriving from specific mechanisms of injury. In conclusion, negativity to FJ-C does not rule out presence of neuronal degeneration and death. Therefore, complementary neuropathological, ultrastructural, and biochemical investigations are essential to identify ongoing neuronal injury and loss.
Supplemental Material
Supplemental Material, Combined_supplemental_materials-Santagostino_et_al - Restricted Sensitivity of FJ-C Staining to Assess Neuronal Degeneration and Death in Preclinical Mouse Studies
Supplemental Material, Combined_supplemental_materials-Santagostino_et_al for Restricted Sensitivity of FJ-C Staining to Assess Neuronal Degeneration and Death in Preclinical Mouse Studies by Sara Francesca Santagostino, Marco Spinazzi and Enrico Radaelli in Veterinary Pathology
Footnotes
Acknowledgments
We would like to thank Dr Bart De Strooper (VIB Center for Brain & Disease Research, Leuven, Belgium, and UK Dementia Research Institute at University College, London, UK) and Dr Fabio Maria Grassi (Institute for Research in Biomedicine, Università della Svizzera Italiana, Bellinzona, Switzerland) for contributing some of the samples included in this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.