
Abstract
Immunohistochemistry (IHC) is a fundamental molecular technique that provides information on protein expression in the context of spatial localization and tissue morphology. IHC is used in all facets of pathology from identifying infectious agents or characterizing tumors in diagnostics, to characterizing cellular and molecular processes in investigative and experimental studies. Confidence in an IHC assay is primarily driven by the degree to which it is validated. There are many approaches to validate an IHC assay’s specificity including bioinformatics approaches using published protein sequences, careful design of positive and negative tissue controls, use of cell pellets with known target protein expression, corroboration of IHC findings with western blots and other analytical methods, and replacement of the primary antibody with an appropriate negative control reagent. Each approach has inherent strengths and weaknesses, and the thoughtful use of these approaches provides cumulative evidence, or a weight of evidence, to support the IHC assay’s specificity and build confidence in a study’s conclusions. Although it is difficult to be 100% confident in the specificity of any IHC assay, it is important to consider how validation approaches provide evidence to support or to question the specificity of labeling, and how that evidence affects the overall interpretation of a study’s results. In this review, we discuss different approaches for IHC antibody validation, with an emphasis on the characterization of antibody specificity in investigative studies. While this review is not prescriptive, it is hoped that it will be thought provoking when considering the interpretation of IHC results.
Immunohistochemistry (IHC) is a standard technique used in all aspects of veterinary pathology. In diagnostics, immunohistochemistry is used to immunophenotype cancers, 2,24,39,40,43,46 provide prognostic information based on protein expression or cell proliferation, 51,57 and identify infectious agents. 34,50 In the context of representative histologic lesions, IHC is often considered a definitive diagnostic test, especially in immunophenotyping cancers and characterizing infectious disease processes. IHC is also used in investigative and toxicology studies to characterize dynamic aspects of disease processes including pathway activation 32,56 and cellular states including cellular activation status, 27 senescence, 26 epithelial to mesenchymal transition, 16 or death, 52 and it is commonly used to characterize the tissue microenvironment 14 and immunologic landscape of pathologic tissues. 21,42 As such, IHC is one of the most common techniques used in studies published in Veterinary Pathology. Although IHC and immunofluorescence are often referred to as “staining” or “immunostaining,” they are much more elegant techniques. Rather than being a stain based on dyes reacting with molecules in tissues, IHC and immunofluorescence rely on antibody binding to specific epitopes. Although epitopes can be carbohydrates 8 or nucleic acids, 59 they are most commonly proteins (which will be the focus of this review). Therefore, IHC and immunofluorescence are antibody-based molecular assays used to define protein expression with the spatial context of the tissue architecture. 35 While this review focuses on chromogenic IHC, considerations on antibody specificity and validation are similar for immunofluorescence assays.
IHC assays are often defined by their sensitivity and specificity. Analytical sensitivity refers to the lower limit of detection of an assay or the minimal amount of protein that can be detected, while analytical specificity refers to the degree to which the antibody only binds the protein of interest. 5 In contrast to analytical definitions, sensitivity and specificity can also be defined in diagnostic contexts. For instance, the likelihood that an animal with an infectious disease will have a “positive” IHC result (diagnostic sensitivity) or the likelihood that a positive cell marker (eg, uroplakin III in urothelial carcinoma) represents a specific tumor type (diagnostic specificity). 39 When considering sensitivity and specificity, it is important to consider the fundamental antibody-epitope reaction that is the basis of IHC. Antibodies bind protein epitopes that are approximately 5 to 6 amino acids in length. These epitopes can have linear or other conformations (based on the proximity of adjacent, noncontinuous amino acids due to protein folding). 35 Epitopes can be specific to a single protein or can be present in multiple proteins, for example, homologous protein family members. 25 Each antibody-epitope interaction has defined affinity and avidity properties. 25,35 Affinity is a measure of the strength of a single antibody-epitope binding interaction, whereas avidity represents the overall strength of antibody binding with consideration of the total antigen-antibody interactions and is influenced by the number of antigen-antibody binding sites. 25 The uniqueness of the epitope, the availability of the epitope in tissue sections (which is influenced by the tissue fixation method), and the antibody’s affinity and avidity to epitopes all influence the sensitivity and specificity of an antibody in an IHC assay. Furthermore, while monoclonal antibodies bind a single epitope, polyclonal antibodies are a combination of antibodies that bind different epitopes on a protein and are more susceptible to lot-to-lot variability, which can further affect the sensitivity and specificity. 25,35 Although antibodies are major determinants of an IHC assay’s sensitivity and specificity, and are the principle discussion point of this article, sensitivity and specificity are also influenced by assay design including pre-analytical tissue handling, fixation, antigen retrieval, and detection methods. 7,35
While western blots and polymerase chain reactions (PCR) use molecular weights or nucleotide lengths, respectively, and/or sequencing to determine the specificity of the detected molecule, similar approaches are not available for IHC. Unanticipated labeling can be specific, revealing new biological insights, or off-target due to labeling of proteins or tissue elements that are not the single protein of interest. The multiple possible underlying causes of nonspecific labeling need to be considered in interpreting IHC labeling. 29,49 This creates the intrinsic challenge of IHC; namely, it is often not possible to definitively prove that an antibody is only binding to the protein of interest, and not to nontarget substances in the tissue sections. To address this issue, the scientific literature, validation studies, and controls are used to generate a body of evidence supporting or refuting the specificity of IHC labeling. 18 The strength of this evidence depends on multiple factors including the depth of target protein characterization (eg, known cellular and tissue expression patterns) and the type, rigor, and results of the validation studies. The strength of the collective body of evidence, also termed the weight of evidence, determines the confidence one has in the specificity of the labeling and therefore the confidence in the results. This evidence is frequently viewed through the independent lenses of the investigator, reviewer, editor, and reader.
Informative reviews on the technical aspects of IHC were recently published in Veterinary Pathology and Toxicologic Pathology. 20,35 Additionally, consensus statements on IHC assay validation in diagnostic veterinary 38 and human pathology, 12 and more recently for multiplexed IHC/immunofluorescence 45 have also been published. The current article by no means replicates or supplants any of the critical information in those publications. Instead, the goal of this article is to discuss different approaches used to characterize the specificity of IHC assays in experimental and diagnostic investigative studies. Each approach has its own advantages and disadvantages (Table 1). While these approaches are not prescriptive, it is hoped that this discussion will enable investigators to consider the strengths and limitations of their antibody validation and the subsequent impact that validation has on the confidence and interpretation of their study in light of the weight of evidence each approach provides.
Comparison of Approaches to Assess IHC Assay Specificity.
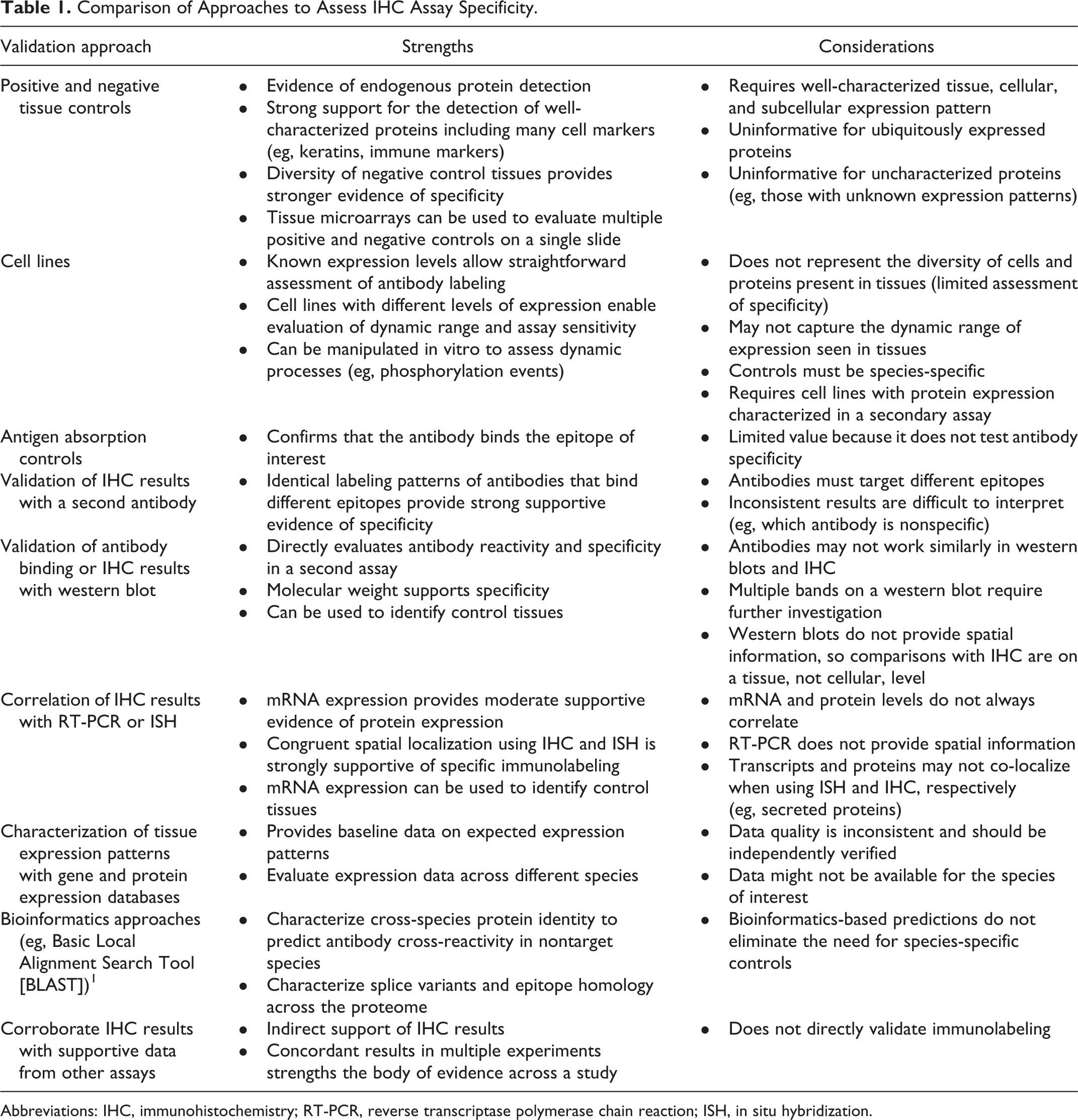
Abbreviations: IHC, immunohistochemistry; RT-PCR, reverse transcriptase polymerase chain reaction; ISH, in situ hybridization.
Validation Studies
Given pathologists’ reliance on IHC, it is important that IHC assays are accurate, reproducible, and reflect true underlying biology. The term validation is relatively broad and can have different connotations in different settings.
Reproducibility
One aspect of assay validation is testing the reproducibility and precision of the assay, frequently on standardized positive and negative controls. Each step in the tissue handling process can significantly affect the quality and immunoreactivity of an IHC assay, including tissue collection, fixation (eg, fixative type, fixation time, fixative to tissue ratio), processing, paraffinization, slide and block storage, and tissue sectioning. The steps in this process, which are commonly termed pre-analytical variables, need to be well controlled and the impact of each step on the final IHC assay should be evaluated. 7,9,58 This may include testing the protocol with different ischemia times (time from sampling to fixation), fixatives, fixation times, unstained slide storage times and conditions, and immunolabeling protocols. This may also include assessments of run-to-run or instrument-to-instrument variability and long-term antibody stability. 12,19,35,38,41,53,54 Ideally, controls used in these studies will have a range of expression levels in order to assess the assay’s sensitivity. 12,38 This validation process defines the conditions under which the assay performs in a reproducible manner and informs the standardized IHC procedure. Consensus statements have been published for diagnostic assay validation in veterinary pathology 38 and human pathology. 12 This type of validation and assay characterization is critical in diagnostic and clinical settings because clinical samples are more likely subjected to pre-analytic variability, and it provides insights in interpreting both the presence and absence of labeling in a patient’s sample.
Sensitivity and Specificity
A second component of IHC validation, which often occurs earlier in the assay development process, is the determination of the sensitivity and specificity of the assay. In diagnostics, these properties are often defined based on diagnostic categories. An example of this is a study that evaluates an antibody in a series of tumors that are expected to express or not express the protein of interest. 4,39,40 These studies define the diagnostic utility and interpretation of an IHC assay; however, they do not always test the specificity of the antibody for its cognate epitope. Frequently, characterization of an antibody’s specificity for its target protein requires additional steps that may or may not be practical given the purpose of the assay. In some cases, such as the diagnostic example above, knowing that an antibody labels a specific tumor type may be sufficient. In other cases, such as characterizing a drug target or the molecular pathogenesis of a disease, more rigorous characterization of the antibody’s specificity may be required. The stringency of antibody validation is inconsistent in both human and veterinary pathology. In published studies, it can range from identifying labeling in areas that are somewhat consistent with the expected results, to validating antibody specificity by using a knock-out animal of the same species as a negative control. The rigor required in primary antibody validation can also vary depending on what is known about the protein’s expression. For well-characterized proteins with distinct expression patterns, normal tissue controls may be sufficient. However, tissue controls are of little value if the antibody detects a ubiquitously expressed protein because it is nearly impossible to determine if the diffuse labeling is specific or nonspecific. Similarly, when the target protein is uncharacterized or when antibodies are used in species that they were not originally developed against, additional steps may be required to confirm specificity. The scientific literature and online gene and protein expression resources provide starting points to understand expected protein expression patterns. However, it is important to remember that the level of data validation in the literature and in online resources is variable, and these resources need to be viewed with a critical eye. It is best to compile information from multiple sources and to independently validate the data in order to prevent being misled.
Extent of Validation
The extent of validation that is needed for an IHC assay is contentious. The only time one can be nearly 100% confident in antibody binding specificity is when labeling is observed in study tissues, but not in tissues from an animal of the same species with deletion or “knock-out” of the gene encoding the target protein. While this approach works well in the mouse, rat, zebrafish, and occasionally in other species, such as the pig or the ferret, knock-out animals from many species are not available. Therefore, demonstrating specificity frequently relies on a weight of evidence approach, 18 and the strength of this evidence is defined by the experiments and controls used in validation studies. At this time, Veterinary Pathology does not have a defined minimal validation standard; instead, each paper is evaluated individually by reviewers and editors to assess the weight of evidence relative to the nature of the study and to be sure the validation methods used in the study are well described. While there are no defined standards, it is important to remember a few basic principles. Experimental design is a key determinant of the quality and validity of an investigative study. A study has stronger supporting evidence and there is more confidence in the study when results are reproducible across large numbers of samples and with multiple complimentary assays, which may include, but are not limited to, reverse transcriptase polymerase chain reaction (RT-PCR), in situ hybridization, flow cytometry, cytokine profiling, or mass spectrometry. 10 The confidence in the results of an assay is defined by the level of validation of the assay. Presented below are approaches that can be used to validate the specificity of an IHC assay. It is important to acknowledge that some of these options are not available in certain contexts due to reagent availability, feasibility, or cost, especially for exotic species. However, the purpose of the discussion is to consider what these approaches can bring to a study by building confidence in the specificity of the IHC results.
Tissue Controls for Antibody Validation
Positive and Negative Control Tissues
One of the most common approaches to validate the specificity of an IHC assay is to use positive and negative control tissues. Positive control tissues are known to express the target protein. Ideally, these tissues have a well-defined pattern of cellular protein expression. Negative controls are tissues that are known not to express the protein of interest. Positive and negative control tissues are frequently used for validating antibodies against cell-specific antigens that have been thoroughly characterized, preferably by a large body of literature. For example, B and T lymphocytes have well-characterized distributions in lymphoid tissues. B cells are concentrated in lymphoid follicles in the spleen and T cells are concentrated in periarteriolar lymphoid sheaths. 6 Since this cellular distribution is so well-defined, normal lymphoid tissues like spleen or lymph node are excellent tissue controls (Fig. 1) and have been used to characterize antibodies against B cell molecules like CD20 and CD19. 15,23 Both of these markers are expressed on the surface of B lymphocytes, and immunolabeling in the lymph node demonstrates distinct membrane labeling of lymphocytes in cortical follicles with limited labeling in the paracortex. 15,23 In this case, the periarteriolar lymphoid sheaths and the paracortex in the spleen and lymph node, respectively, can serve as a negative control because few B cells expressing these proteins should be present in these regions. However, it is important to remember that the specificity of an antibody is only validated to the degree that negative control tissues are represented in the validation study. Each tissue has a unique proteome, and lack of off-target binding in one tissue does not eliminate the chance for off-target binding in other tissues. Therefore, the more varied the negative control tissues used in a validation study, the more confidence one has that the antibody does not bind non-specifically in other tissues.
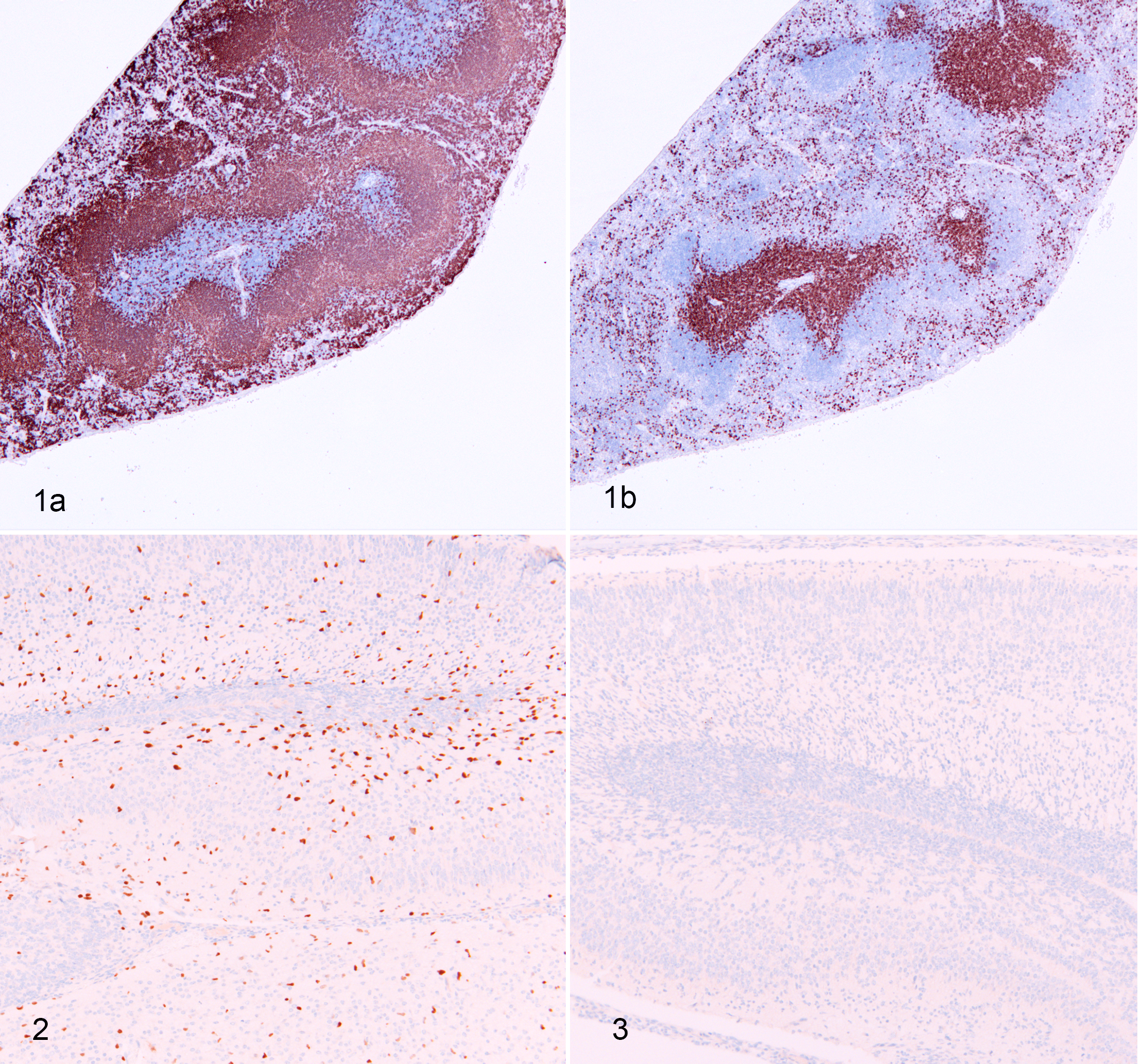
Normal spleen, wild-type mouse. For antigens expressed in known areas within a tissue, the location of immunolabeling (and lack of immunolabeling) can help validate the reactivity and specificity of the antibody. (a) B220-positive B lymphocytes are present in lymphoid follicles and (b) CD3-positive lymphocytes are present in periarteriolar lymphoid sheaths. Immunohistochemistry for B220 (a) and CD3 (b).
The use of tissue controls requires a baseline knowledge of the expected tissue, including the cellular and subcellular (nucleus, cytoplasm, or membrane), localization of the protein, as well as knowledge of where the protein should not be expressed. 38 Although validation with tissue controls is often suitable for well-characterized proteins, it is important to think about what these controls do and do not reveal about the antibody’s specificity. These controls show that the antibody binds to tissues, cells, and subcellular compartments in a manner that is compatible with the known expression pattern of the protein. However, these controls do not differentiate whether the antibody is specifically binding to the protein of interest or a related protein that is expressed in the same location. This highlights 2 important principles: (1) all IHC interpretation is based on a weight of evidence and (2) the more supporting data that are available to confirm the expected pattern of protein expression, the more confidence one will have in the IHC interpretation. This may require further validation with complementary assays, as described below.
Antibody Concentration
If a protein is expressed at varying levels in different cell types, evaluating a variety of tissues during the validation process also helps characterize the dynamic range of the assay. This is optimally done in combination with a primary antibody dilution series to determine the best signal to noise ratio of the assay and identify the sensitivity, or the lower limit of protein detection in the assay. This is an important consideration because the absence of labeling may not mean absence of protein; the protein may just be expressed below the assay’s limit of detection. Frequently, there is a trade-off between the sensitivity and specificity of an assay. The optimal signal to noise ratio is often determined empirically based on the needs of the assay (high sensitivity vs high specificity) and the interpretability of the assay (eg, the ability to differentiate signal from noise). Tissue microarrays that contain cores from multiple different tissues on a single slide provide a high throughput method to validate against a spectrum of control tissues. 35
Species-Specificity
Veterinary pathologists deal with a variety of animals, and there are few species-specific antibodies available for many of them. Because of this, investigators frequently need to use antibodies developed against human or mouse proteins for their studies. 35 While a manufacturer’s datasheet can provide some information about the expected cross-species reactivity, this information also needs to be interpreted based on the quality of the data. 5 Protein sequence alignments can provide information on the likelihood of cross-species immunoreactivity, especially if the cognate epitope is known. 5 This approach was used in evaluating an anti-human CD30 antibody in feline tissues. 4 Unfortunately, an antibody that works well in one species, will not always perform similarly in a different species. 35 An example of this is melan A, which is a commonly used marker for canine melanoma. 36,43 When an established anti-canine melan A IHC protocol was evaluated in a series of equine melanomas, none of the tumors had detectable labeling. 37 This highlights the fact that an established IHC protocol for one species will not always behave similarly for a second species; therefore, control tissues should be from the species that is being evaluated in the study. This may be straightforward for well-defined cell markers; however, not all species will have identical protein expression patterns. For instance, genes encoding receptor interacting protein kinase 3 (RIPK3) and mixed lineage kinase like (MLKL), the effectors of necroptosis, are absent from marsupial genomes. 31 MLKL is also absent in carnivores, while RIPK3 is absent in birds but MLKL is present. 31 This reiterates the importance of building evidence for the appropriateness of a control, and in some cases, this may require additional legwork to confirm that the species even expresses the protein.
For mice and some other laboratory species, tissues from knock-out animals can serve as optimal negative controls, although there are several caveats to this approach as well. 55 In an ideal situation, a germline knock-out mouse is engineered so that there is no mRNA or protein expression. In this case, wild-type and knock-out tissues can be evaluated in parallel, demonstrating specific labeling in the wild-type tissue and no immunolabeling in the knock-out tissue, thereby confirming the specificity. 55 An ideal example of this is Olig2 IHC in the Olig2 knock-out mouse (Figs. 2, 3). Distinct labeling of oligodendrocytes was observed in the brain of the wild-type mouse, while no labeling was present in the knock-out mouse. Unfortunately, in some cases, a knock-out mouse may lack functional protein, but may still produce a truncated product that can be detected by an antibody. An example of this was an Etv5-/- mouse that expressed increased amounts of a nonfunctional protein due to the absence of a negative feedback loop to control transcription. In this case, ETV5 labeling still occurred in the tissue section even though functional ETV5 protein was absent. 30 When using an inducible system to delete a gene in an adult animal, such as a tamoxifen-inducible cre.ERT2 mouse, the efficiency of gene deletion is tissue and cell-specific and can be incomplete, so these control tissues may need to be validated via PCR or western blotting prior to being used as controls. 55
Identification of Infectious Agents
Control tissues for IHC-based detection of infectious agents have unique considerations. The presence of the infectious agent in the control tissues should be confirmed by an alternative method (eg, culture, PCR-based detection, in situ hybridization [ISH]) and the tissues should demonstrate characteristic histologic lesions of the infectious agent. 35 For example, immunolabeling of Lawsonia intracellularis in equine tissue is identified in hyperplastic and tortuous intestinal crypts and highlights bacteria with characteristic spiral morphologies (Fig. 4). A second example is the identification of bovid herpesvirus-1 immunolabeling in a characteristic lytic lesion in the liver of a calf (Fig. 5). Some antibodies will cross-react with related infectious agents. For example, a polyclonal anti–Neospora caninum antibody can also detect Sarcocystis sp. in bovine heart. 48 Sometimes this is advantageous in detecting related infectious agents in different species. The same antibody can be used to detect coronavirus in pigs with transmissible gastroenteritis and cats with feline infectious peritonitis. 54 The fact that an antibody can detect different related organisms does not preclude the use of the antibody, but it does affect how the IHC result is interpreted. Testing the antibody on tissues with related infectious agents, especially agents that cause morphologically similar lesions, is important to understand the specificity of the antibody. Ultimately, the specificity is defined by the diversity of infectious agents that the antibody is tested against, so broader interrogation of tissues infected with related microbes will provide a better understanding of the selectivity of the antibody and confidence of the IHC results.
Lawsonia intracellularis infection, small intestine, horse. There is immunolabeling of intracellular spirochetes within hyperplastic intestinal crypts. The location and morphology of the immunolabeling aids in the validation of the assay. Immunohistochemistry for Lawsonia intracellularis with DAB chromogen and hematoxylin counterstain.
Cell Line Controls
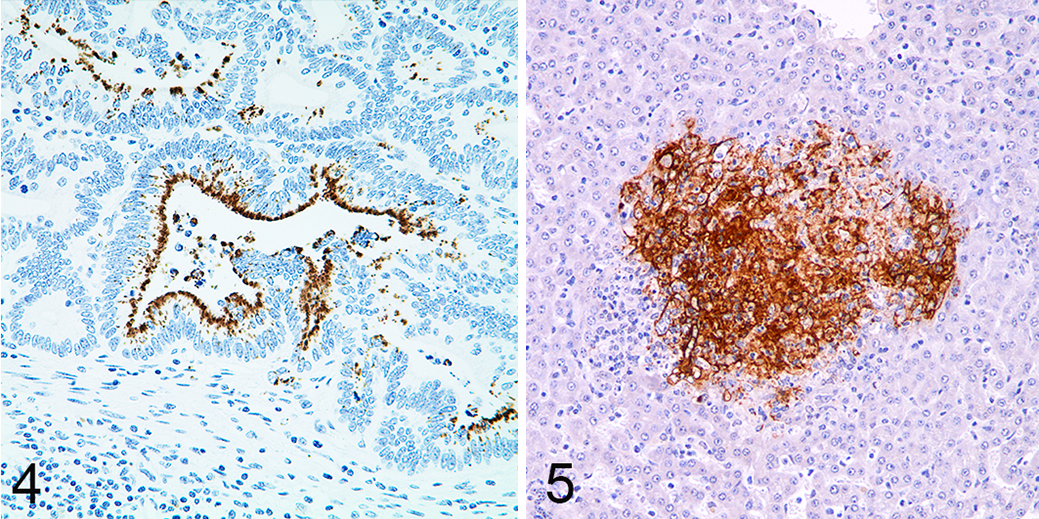
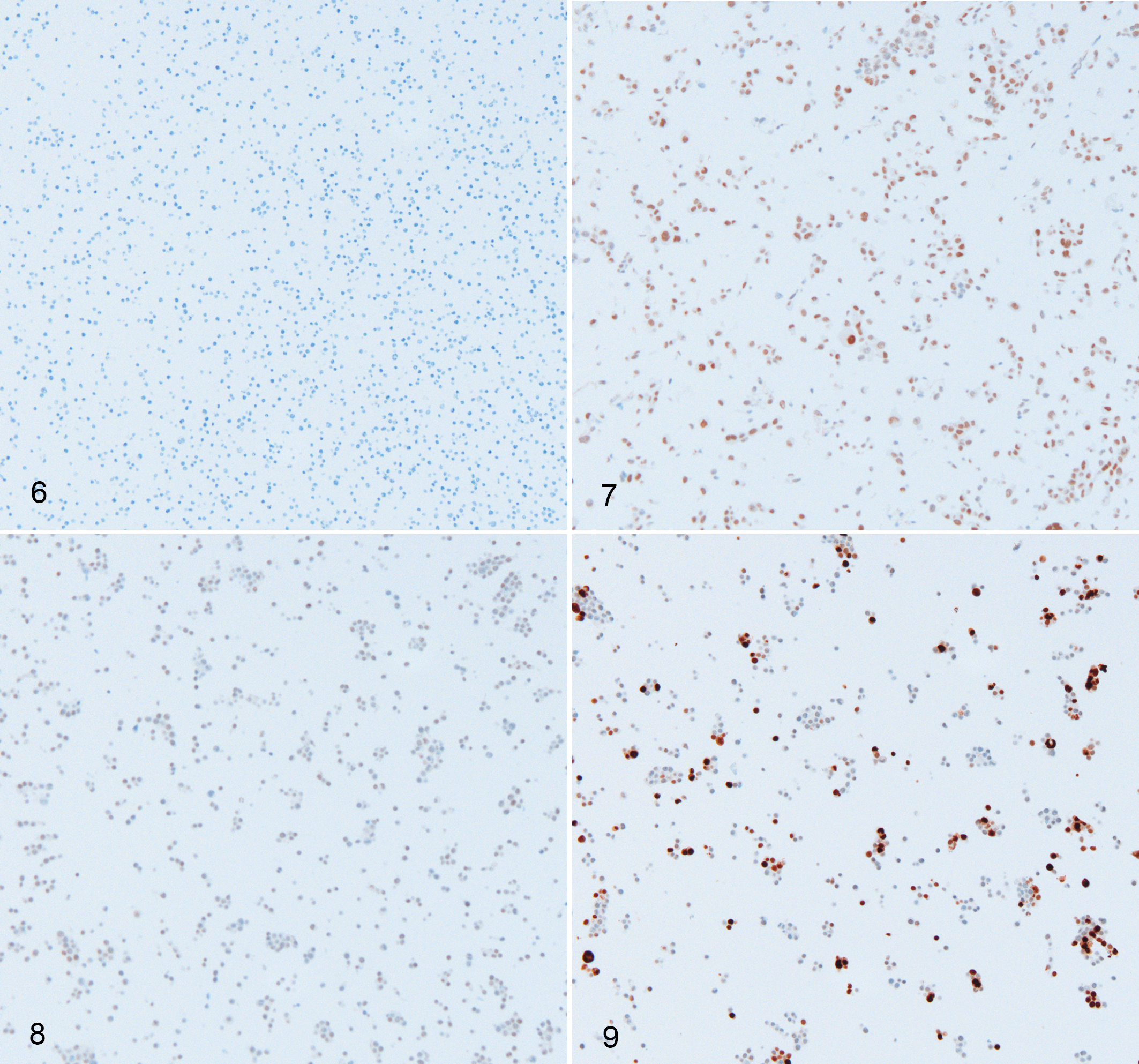
Cell line controls offer a versatile option when tissue controls are not available or are inadequate. There are 2 basic categories of cell line controls: (1) endogenous cell lines and (2) engineered cell lines. Endogenous cell lines primarily consist of cancer cells and other cell populations that can be grown in culture, like fibroblasts or bone marrow–derived macrophages, that have known levels of target protein expression and have not been manipulated to overexpress the protein. Ideally, cancer cell lines with high, medium, and weak/absent protein expression can be identified in order to demonstrate the dynamic range of immunolabeling and assess the assay sensitivity (Figs. 6, 7). A challenge with endogenous cell lines is that they need to be from the target species to ensure antigen specificity. While there are numerous human cancer lines, there are fewer options for other species; although increasing numbers of canine cancer cell lines are being developed and characterized. 13,17,44 Engineered cell lines can be made by overexpressing a protein of interest to create a positive control (Figs. 8, 9), or by deleting the gene of interest in order to make a negative control. CRISPR/Cas9 gene editing tools have significantly increased the feasibility of generating knock-out cell lines. 22 In either case, the wild-type cell line can serve as the inverse control (eg, negative control for the overexpressing cell line or positive control for the knock-out or knock-down cell line). While knock-out cell lines should be from the species of interest, overexpressing cell lines can be from a different species if the protein being overexpressed is from the target species. Cell line controls should be fixed, processed, and embedded in a similar manner as the study tissues. If the study tissues are paraffin-embedded, the cell lines should be expanded, pelleted, fixed in formalin, and routinely processed and embedded in paraffin.
Cell pellets, human. Immunolabeling for an undisclosed protein. Antibody sensitivity and specificity is tested using cells known to express low or high levels of the protein, and by comparing a cell line that is non-transfected to one that is transfected to overexpress the protein.
Cell lines provide versatility because they can be subjected to experimental conditions prior to collection. This is helpful when controls are needed to assess signaling pathway activation. For instance, cells can be stimulated with cytokines to assess downstream pathway activation with phospho-epitope-specific antibodies. In this scenario, the stimulated cells serve as a positive control while the unstimulated cells serve as a negative control. Additionally, in the case of phospho-epitope IHC assays, pretreating the slides with phosphatase can serve as an added control to test for phospho-specificity. Following phosphatase treatment, the phospho-epitope should be degraded and no longer available for binding, whereas labeling should still be evident in the untreated sample (Figs. 10–12).
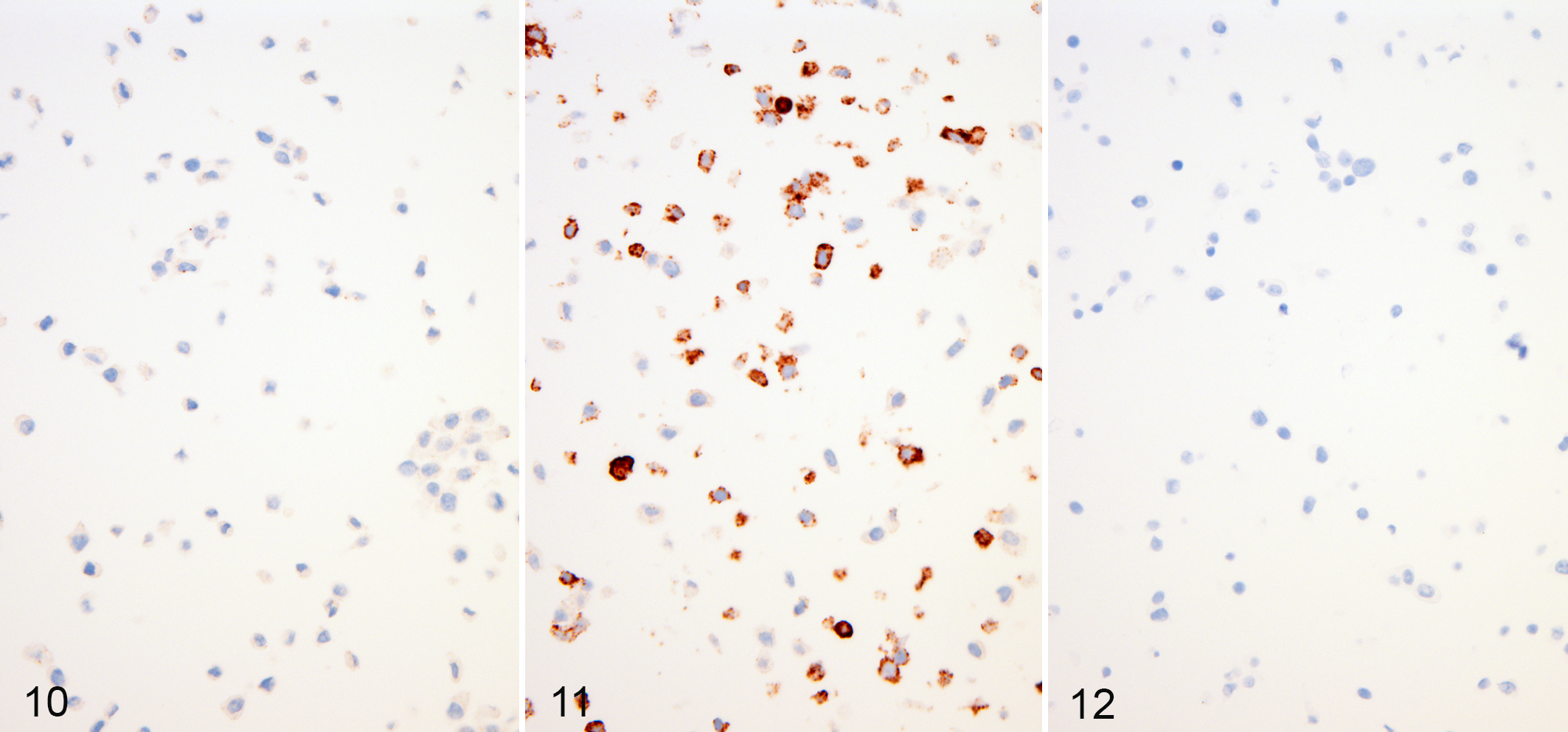
L929 fibroblast cells, mouse. Phospho-RIPK3 immunohistochemistry. In vitro treatment of cell lines with agonists validates the immunohistochemistry data.
While cell line controls have their advantages, including the confirmation that the protein is or is not expressed in the cells, they also have several limitations. First, cell line controls may lack dynamic range. 18 In particular, overexpressing cell lines frequently express proteins at levels that far exceed endogenous protein expression (Figs. 8, 9). Therefore, while these cell lines are helpful to identify antibodies that detect the protein of interest, they may lead one to overestimate the sensitivity of the assay. If overexpressing, knock-out, or knock-down approaches are used to generate control cell lines, the efficiency of the genetic manipulation needs to be considered when interpreting the controls. Unless the cells are developed from single-cell clones, the genetic change might not be equal in all cells. In overexpressing and knock-out lines this might result in a mosaic of cells that do and do not have any labeling (Figs. 8, 9). In cells generated using shRNA to knock-down a target mRNA, this might result in reduced, but not absent signal in cells. The true Achilles’ heel of using cell line controls is that their proteome only reflects a single cell and, therefore, might not be representative of the tissue of interest. Tissues are composed of many different cell types that each have their own proteome or protein expression profile. Unanticipated antibody binding might be due to the expression of a homologous protein in a specific cell type or due to nonspecific binding in a specific cell type. 25,29 Unfortunately, this diversity of protein expression cannot be evaluated in cell line controls because the cell lines intrinsically only represent a single cell type. Therefore, while cell lines are suitable for showing the antibody detects the protein of interest, they are more limited in showing that the antibody does not detect other proteins.
Synthetic Antigen Controls
Synthetic antigen controls offer a novel alternative to cell pellet controls. These controls are made by forming formalin cross-linked gels that contain a defined concentration of small peptides, protein domains, or full-length proteins in combination with a protein matrix, such as bovine serum albumin. Since the protein concentration of these gels can be controlled, they offer the opportunity of being standardized controls for assessing sensitivity and dynamic range and can serve as intrarun and interinstitution quality controls. However, the principal disadvantage of these controls is that they lack the antigenic diversity of tissues or their cellular compartmentalization that may aid in interpretation. 19
Study Controls
Aside from initially validating an IHC assay, positive and negative controls should be included in every IHC run to ensure the adequate assay performance. 18,35,38 These controls confirm that the primary antibody is labeling the tissues in a consistent manner. A second type of negative control is used to determine if any signal is being produced due to nonspecific labeling of the secondary antibody or the detection system. 18,29,35 This control is based on running the assay without the primary antibody. Simply leaving out the primary antibody or replacing the primary antibody with buffer is considered an insufficient negative control; instead, it is preferable to replace the primary antibody with an antibody of the identical isotype. 18 Isotype-specific monoclonal and polyclonal antibodies are available commercially for use as negative controls. Alternatively, antibodies of matching isotypes that do not bind in a similar manner as the primary antibody of interest can be used as a negative control. Pre-immune sera can also serve as a negative control for polyclonal antibodies. Replacing the primary antibody with buffer tests nonspecific labeling due to the detection system (secondary antibody, streptavidin/biotin or polymer, chromogen), while replacing the primary antibody with an isotype-matched antibody additionally assesses for nonspecific binding that is dependent on the Fc portion of the antibody. 18,35 While these negative controls are important, they do not assess all potential causes of nonspecific labeling.
Cross-Validation: Using Different Assays to Validate IHC Data
The approaches described above represent scenarios where control tissues or cell lines can be identified. However, these controls are not always available or known, and even so, they may have gaps in their ability to fully evaluate the specificity of an assay. In these cases, complementary assays can be employed to strengthen the case for specificity. 10 We often think of this in terms of identifying controls and initial IHC validation; however, IHC results can be also be supported by complementary methods or data. 5 For example, detection of the protein or encoding transcript with a second assay provides further validation of the IHC results. This can be particularly important if the protein is not well characterized.
Absorption Controls
Absorption controls are used to test if the primary antibody binds the antigen of interest. In this method, the antibody is first incubated overnight with the antigen. Following incubation, the antibody should no longer be able to label tissues when it is included in the IHC reaction due to saturated binding of the antigen. It is important to keep in mind that the absorption control will work better if the antigen is a purified peptide. If the antigen is a whole protein, nonspecific binding might take place between the protein and the tissue, leading to incorrect interpretations. While this approach tests the ability of the antibody to bind the target protein, it provides no information as to the specificity of this binding, since no competing epitopes are being tested. As such, this approach has no value in routine validation studies. 18
Validation With a Second Antibody
The use of a second antibody that detects a different epitope on the target protein can provide evidence that IHC labeling pattern is specific. 5 In principle, if both antibodies are specific, they should have identical labeling patterns despite detecting different epitopes. However, this approach has theoretical complications if one epitope is unavailable in select cell populations, perhaps due to being engaged in protein complexes. Additionally, this approach does not work as well for polyclonal antibodies or if 2 antibodies detect the same epitope because this approach will not be able to identify promiscuous binding due to homologous amino acid sequences in other proteins.
Western Blot
Western blotting is a preferred method to validate IHC antibodies because the antibody used in the IHC assay can be directly tested and the specificity of detection can be determined based on the molecular weight of the detected protein. 5,18,35 Ideally, the antibody will detect a single band at the expected molecular weight; however, more detective work is needed if multiple bands are present on the blot. 5 Detection of additional bands on the western blot could represent the normal biology of the target protein; for instance, posttranslational modifications, splice variants, or cleavage products. 28 Alternatively, the additional bands could indicate antibody binding to nontarget proteins. Mass spectrometry of individual bands might be required to determine the identity of these bands, especially if the protein is not well characterized. 3
Despite the great potential western blots have to validate IHC antibodies, one should remember that antibodies may behave differently in IHC versus western blot. While IHC often utilizes formalin-fixed, paraffin-embedded (FFPE) tissues, fresh or frozen (nonfixed) tissues are often used for western blots. In order to separate proteins based on their molecular weight on a western blot, proteins are denatured and separated by gel electrophoresis. 18,25 In contrast, formalin crosslinks proteins. This difference in protein states may significantly affect the detection of epitopes in one assay versus the other. 35 For instance, epitopes that are detected via western blots could be hidden following formalin fixation; or if the epitope is reliant on the protein’s tertiary structure, this could be lost when the protein is denatured. Confirming specificity in a western blot is valuable supportive evidence, but it does not prove that it is specific in IHC assays. Likewise, absence of specific bands in western blots does not prove an inability of the antibody to detect the epitope of interest in IHC.
While western blots can be used to directly confirm that the IHC antibody specifically detects the protein of interest and does not detect other proteins, they can also be used to identify appropriate controls or to directly validate the results of a study. Western blots often include a loading control protein for normalization, which provides the ability to qualitatively assess the level of protein expression. This is valuable for identifying high, medium, and low expressing controls, and for comparing expression levels between tissue samples. However, western blots do not provide any spatial information, so direct comparisons between IHC and western blot data can be challenging in complex tissues. Additionally, fresh or fresh frozen tissues are often preferred for western blots, which might not be available especially in retrospective studies.
Reverse Transcriptase PCR and In Situ Hybridization
RT-PCR and ISH are methods for assessing RNA expression. While RNA and protein levels do not necessarily correlate, at minimum, RNA levels can serve as a surrogate assessment of whether the protein is or is not expressed in the tissue of interest, even if it cannot predict the level of expression. Both methods can be used with FFPE tissues, although the use of FFPE tissues requires specific considerations for assay development, particularly for PCR assays. 11 While PCR has the advantage that it can be quantitative, it lacks the spatial data provided by IHC. A combination of IHC, western blotting, and RT-PCR was used to confirm insulin-like growth factor 2 (IGF-2) expression in a pulmonary adenocarcinoma associated with hypoglycemia. 33 The authors further assessed insulin-like growth factor 1 (IGF-1) with each of these assays. By using 3 techniques to characterize IGF-2 expression and taking similar steps to rule out IGF-1, the authors were able to provide robust, nearly irrefutable evidence that tumor-derived IGF-2 was the cause of the hypoglycemia. 33
In contrast to RT-PCR, ISH provides spatial information that can be directly correlated with IHC data. An example of this is the detection of West Nile virus in horses by IHC and ISH. The correlation of labeling between the 2 assays adds confidence in the specificity of the labeling. 47 Similarly, detection of ETV1 protein in the mouse cerebral cortex by IHC and transcript by ISH increases the confidence in each individual assay (Fig. 13). However, it is good to remember that for secreted proteins, the protein and RNA may localize to different cells: while the RNA is produced in one cell, the protein product may be secreted and bind to a different cell or be present in the extracellular matrix. Additionally, transcript and protein levels do not always correlate because of the rate of transcription, translation, or protein turnover. While congruent IHC and ISH results add confidence, differing results may need to be investigated further to determine if this difference is due to analytical or biological factors. Finally, if the goal of a study is to detect posttranslational modifications, like phosphorylation events, detection of RNA provides only limited data in validating the specificity of the IHC assay.
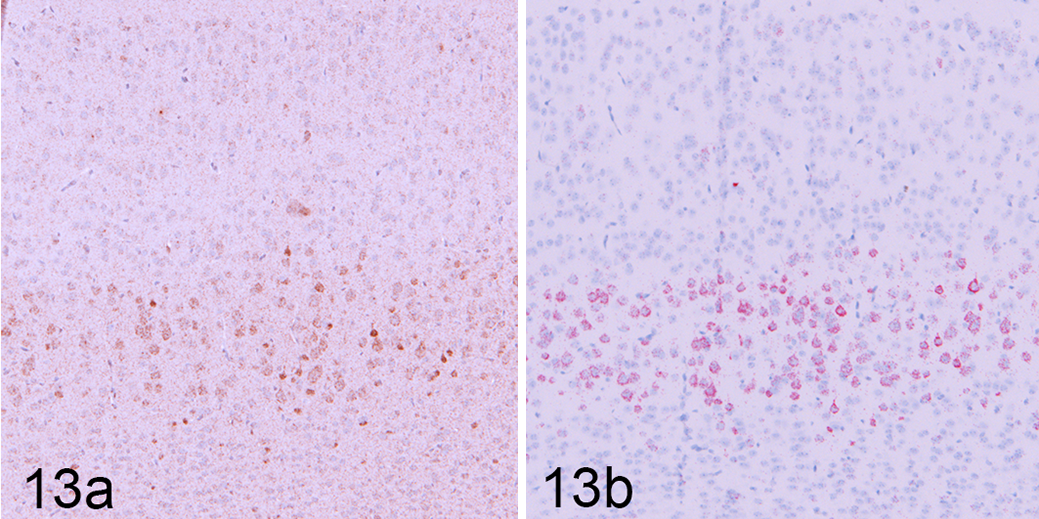
Normal brain, mouse. (a) ETV1 immunolabeling in the cerebral cortex. (b) In situ hybridization labeling of Etv1 transcript in the cerebral cortex. Co-localization of ETV1 protein and Etv1 transcript in these 2 assays builds confidence in the specific nuclear labeling of the ETV1 IHC, despite an increased amount of background.
Supporting IHC Results With Complementary Data
Beyond corroborating an IHC labeling pattern with a secondary assay, a study’s results can be supported by integrating data using multiple assays and measures. 5 This is especially important in mechanistic studies where one is trying to understand disease pathogenesis. For instance, if the goal of a mechanistic cancer study is to show mitogen-activated protein kinase (MAPK) pathway activation in a given type of cancer, one can use IHC to detect phospho-ERK in the tissues. However, these data can be further strengthened by showing gene expression changes in the cancer that are reflective of changes in MAPK signaling, as well as in vitro experiments demonstrating that cancer cell lines respond to MAPK inhibitors. Similarly, while IHC can provide critical data characterizing immune infiltrates in infectious or immune-mediated disease, flow cytometric analysis of the primary lesions or draining lymphoid tissues and gene expression data can further support the IHC results. In studying histologic changes in canine dilated cardiomyopathy, a combination of morphometry, IHC, and PCR was used to develop a more complete picture of tissue remodeling. 14 While these ancillary assays and measurements do not provide direct support for an IHC assay, they do add to the weight of evidence and therefore the confidence in a study’s conclusions.
Conclusions
IHC is fundamental to modern pathology. It is the primary molecular assay performed by pathologists, and pathologists are the standard bearers for using this assay. As such, it is our duty to ensure the results derived from this assay are of the highest possible quality. The approaches described in this manuscript are not prescriptive. Instead, they represent different approaches to validating the specificity of an IHC assay and highlight special considerations associated with each approach. The primary message of this article is that IHC validation is a weight-of-evidence process. Some approaches provide only weak evidence of antibody specificity, while others provide strong or nearly irrefutable evidence. The strength of evidence provided may be context-specific. The ability to build the case that IHC labeling is specific depends on how well characterized the protein is, how discretely the protein is expressed (a well-defined localization vs global expression), the target species, and the available resources. Additionally, the quality of an IHC assay will always be determined by the antigenicity of the protein and the quality of the primary antibody. However, it is critical to remember that the more robust the validation process is, the more confidence one will have in the results.
Footnotes
Acknowledgements
We would like to acknowledge our colleagues who allowed us to use images from collaborations, specifically Kim Newton and Anwesha Dey, as well as the Genentech IHC/ISH Laboratory, including Debra Dunlap, Felix Chu, and Susan Sa, and the Purdue University Animal Disease Diagnostic Laboratory, including Dee DuSold, who assisted in the original IHC/ISH depicted in the figures.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Joshua D. Webster and Margaret Solon are employees of Genentech and Roche stockholders. Katherine N. Gibson-Corley has no conflicts of interest to disclose.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.