
Abstract
A 4-month-old female mixed-breed cat showed gait disturbance and eventual dysstasia with intention tremor and died at 14 months of age. Postmortem histological analysis revealed degeneration of neuronal cells, alveolar epithelial cells, hepatocytes, and renal tubular epithelial cells. Infiltration of macrophages was observed in the nervous system and visceral organs. The cytoplasm of neuronal cells was filled with Luxol fast blue (LFB)-negative and periodic acid-Schiff (PAS)-negative granules, and the cytoplasm of macrophages was LFB-positive and PAS-negative. Ultrastructurally, concentric deposits were observed in the brain and visceral organs. Genetic and biochemical analysis revealed a nonsense mutation (c.1017G>A) in the SMPD1 gene, a decrease of SMPD1 mRNA expression, and reduced acid sphingomyelinase immunoreactivity. Therefore, this cat was diagnosed as having Niemann-Pick disease with a mutation in the SMPD1 gene, a syndrome analogous to human Niemann-Pick disease type A.
Niemann-Pick disease (NPD) is a fatal autosomal recessive lysosomal disease characterized by the accumulation of sphingomyelin and cholesterol in the lysosomes. In humans, NPD is classified into 5 variants (types A–E) according to clinical and genetic findings. Types A, B, and E NPD are caused by a deficient activity of the enzyme acid sphingomyelinase (ASM), encoded by the sphingomyelin phosphodiesterase 1 (SMPD1) gene. Types C and D NPD are caused by abnormal NPC1 or NPC2 protein, encoded by the NPC1 or NPC2 gene, respectively. Type A NPD is a fatal early-onset lysosomal storage disease leading to death within first 3 years of life. 7
Type C variant with the mutation of NPC1 10,14 and NPC2 genes 17 has also been reported in cats. In addition, feline type C NPD with a missense mutation of the NPC1 gene (c.2864G>C) has been maintained and studied as an animal model of human type C NPD. 15 A syndrome analogous to type A NPD has also been reported in Siamese cats, 4,6,13,16 a domestic shorthair cat, 12 and a Balinese cat 2 by detecting ASM deficiency. However, a genetic mutation in the SMPD1 gene has never been reported in cats. Here, we describe a kitten diagnosed as type A NPD with a mutation in the SMPD1 gene.
A 4-month-old female mixed-breed cat (weight at 1.8 kg) showed weakness and gait disturbance and was referred to a veterinary hospital. On neurological examination at the first presentation, deficits of proprioception and loss of spinal reflexes for all limbs were observed, while superficial pain was preserved. Blood tests including titers to FIV (feline immunodeficiency virus), FeLV (feline leukemia virus), and FCoV (feline coronavirus) were within the reference ranges. The cat was treated with prednisolone for 1 week, but there was no improvement. Then clinical signs progressed gradually to exhibit dysstasia and intention tremor. Mentation and appetite remained normal until the end stage. The cat died at 14 months of age, and necropsy was performed. On gross examination, the brain was edematous, and the liver was enlarged and pale. The cat had a littermate that showed the same neurologic signs and died; however, a necropsy was not performed.
For histopathology, samples of nervous tissues and major visceral organs were fixed in 10% neutral-buffered formalin, and embedded in paraffin wax. Formalin-fixed and paraffin-embedded tissues were sectioned at 4 µm thickness, and stained with hematoxylin and eosin (HE), Luxol fast blue (LFB), periodic acid-Schiff (PAS), and Alcian blue (pH 4.1). Serial sections were also subjected to immunohistochemical analysis using the Envision polymer (Dako, Glostrup, Denmark) and 3,3′-diaminobenzidine tetrahydrochloride as a chromogen. The following primary antibodies were used: mouse anti-MAP2 (1:200, Millipore, Temecula, CA), rabbit anti-Iba-1 (1:300, WAKO, Osaka, Japan), and anti-ASM (1:100, LSBio, Seattle, WA). For antigen retrieval, the sections were treated at 121°C for 10 minutes in citrate buffer (pH 6.0). For ultrastructural examination, samples of formalin-fixed cerebral cortex, liver, and spleen were washed with phosphate-buffered saline (pH 7.4) and then post-fixed in 1% osmium tetroxide. Samples were dehydrated and embedded in resin Luveak-812 (Nacalai Tesque, Kyoto, Japan). Uranyl acetate and lead nitrate–stained ultrathin sections were then examined with a transmission electron microscope (JEM-1400Plus, JEOL, Tokyo, Japan). For molecular analysis of the SMPD1 gene, genomic DNA and total RNA were extracted from the frozen brain tissue using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) and RNeasy Mini Kit (Qiagen), respectively. Extracted total RNA was used for cDNA synthesis (Prime Script RT-PCR kit, TaKaRa, Shiga, Japan). The purified DNA and cDNA were used for subsequent PCRs (polymerase chain reactions). Three feline SMPD1 gene-specific PCR primer pairs were used: primer pair 1, which amplifies a 999 bp fragment that incorporates exon 1 and exon 2 of the SMPD1 gene, 5′-CGGATCCCGGCTGCTG-3′ (forward) and 5′-CGGCCCCTTTGATATGGTGT-3′ (reverse); primer pair 2, which amplifies a 388 bp fragment that incorporates exon 2 and exon 3 of the SMPD1 gene, 5′-TGGATACTGGGGCGAGTACA-3′ (forward) and 5′-GGAAAGGGCATAGAACCCCC-3′ (reverse); and primer pair 3, which amplifies a 564 bp fragment that incorporates exon 6 of the SMPD1 gene, 5′-GAGCTCGAGAAACCTACGGG-3′ (forward) and 5′-GTGAGAGCTCCTGTGTGGTC-3′ (reverse). PCR was performed as follows: 30 cycles at 98°C for 10 seconds, 60°C for 30 seconds. The PCR products were submitted for sequence analysis (FASMAC, Toyama, Japan). PCR was performed in duplicate, and an age-matched unaffected cat was used for confirming normal sequences of the feline SMPD1 gene. For western blotting, brain samples were homogenized in 7.5 volumes of TBS buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EGTA, and 1 mM EDTA. After centrifugation at 23 000 rpm at 4°C for 15 minutes, the supernatants were collected and dissolved in Laemmli Sample Buffer (SB), which included 2-mercaptoethanol, and boiled for 10 minutes. For protein analysis, the proteins dissolved in Laemmli SB were separated using a 5% to 20% gradient polyacrylamide gel (ATTO, Tokyo, Japan), and then transferred to a 0.20-μm-thick nitrocellulose membrane (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Nonspecific binding was blocked by treatment with 1% skim milk for 60 minutes. The membranes were probed with the following antibodies at 4°C overnight: rabbit anti-ASM (1:500, LSBio) and horseradish peroxidase (HRP)-conjugated mouse anti-β-actin (clone 8H10D10, 1:5000, Cell Signaling Technology, Beverly, MA). After washing the membranes with TBS containing Tween 20, HRP-conjugated sheep anti-rabbit IgG antibody (1:5000, GE Healthcare UK, Little Chalfont, UK) was applied. Blots were developed using ECL Select Western Blotting Detection Reagent (GE Healthcare Bio-Sciences AB). Immunoreactive bands were detected using the ChemiDoc XRS+ System (Bio-Rad Laboratories, Hercules, CA).
Histologically, degeneration of neuronal cells characterized by the swollen perikaryon and infiltration of foamy macrophages were observed throughout the central nervous system (CNS; Fig. 1 and Supplemental Figs. S1, S2). These cells contained numerous, weakly eosinophilic granules in the cytoplasm. The cerebral cortex and Ammon’s horn of the hippocampus were severely affected, but the white matter and dentate gyrus of the hippocampus were less affected. In the cerebellum, Purkinje cells were mildly affected and reduced in number. Degenerated neuronal cells were also observed in the brainstem including the midbrain, pons and medulla, and spinal cord. Mild axonal degeneration characterized by swollen and fragmented axons was also observed. Moderate astrocytosis was observed in the affected areas. The cytoplasmic granules in the neuronal cells were negative for LFB, PAS, and Alcian blue, while granules in the macrophages in the lesions were positive for LFB, but negative for PAS and Alcian blue (Figs. 2, 3). Ultrastructural examination revealed the accumulation of variably sized and various numbers of membranous concentric bodies in the cytoplasm of the affected cells in the brain (Fig. 4).
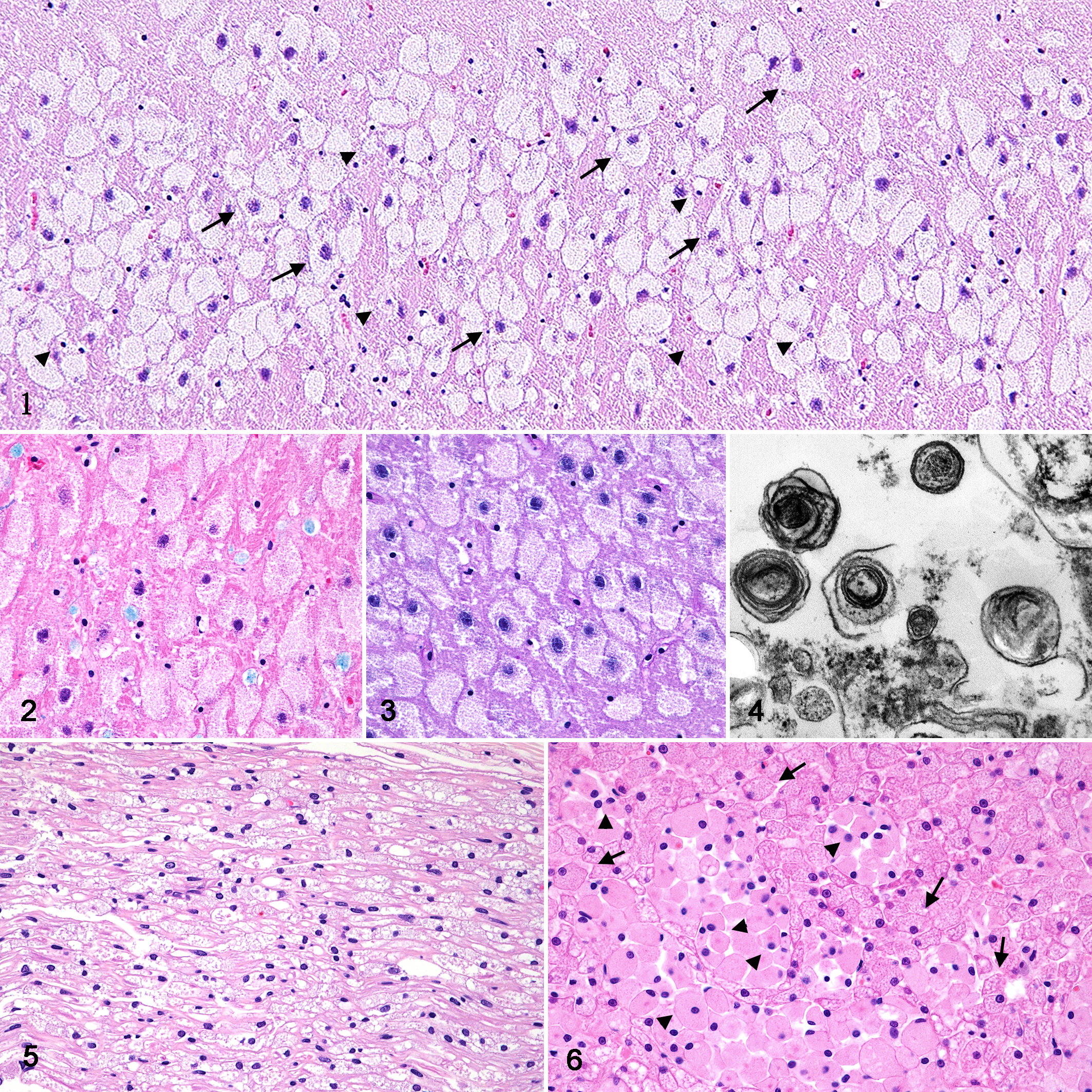
In the peripheral nerves system (PNS), the trigeminal nerve, dorsal and ventral roots of the spinal cord, ganglion of the heart atrium, and nerve plexus of the small intestines were affected with degeneration of neuronal cells and mild infiltration of macrophages. In the ischiatic nerve and branchial plexus, mild myelin loss and swollen axons with intracytoplasmic granules were observed (Fig. 5).
In the visceral organs, multifocal aggregates of macrophages were observed in the spleen, lungs, lamina propria of the stomach, small and large intestines, liver, pancreas, kidneys, and adrenal glands. The alveolar epithelial cells, hepatocytes, and renal tubular epithelial cells were also degenerated with the accumulation of cytoplasmic granules (Fig. 6, Supplemental Figs. S3–S8). Ultrastructural examination revealed the accumulation of stacked membranous and occasionally concentric materials in the cytoplasm of hepatocytes (Supplemental Fig. S9). Also, variably sized membranous concentric bodies were found in the cytoplasm of macrophages in the liver and spleen (Supplemental Figs. S10, S11). The granules within the macrophages and epithelial cells of the visceral organs were positive for LFB.
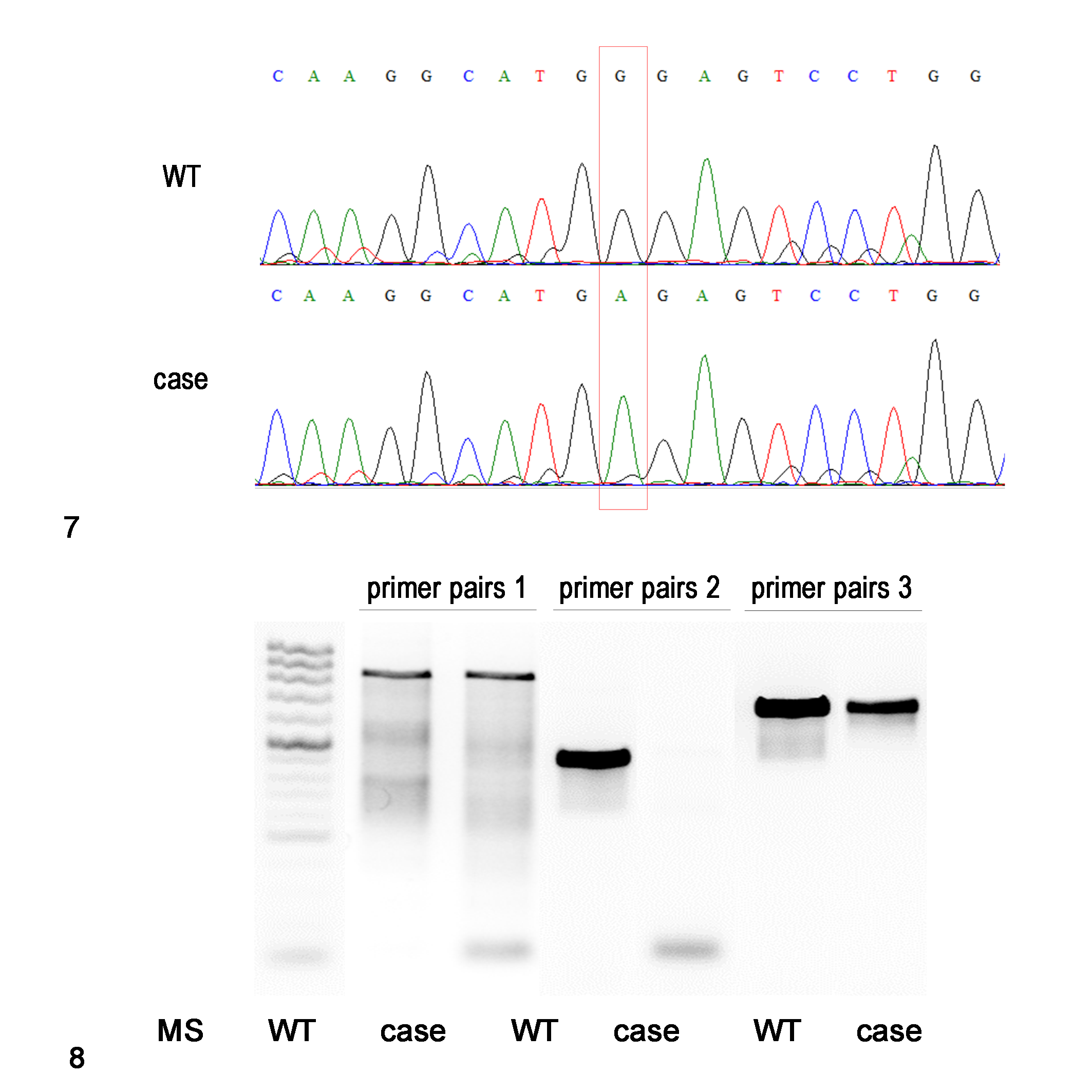
DNA sequencing analysis identified the c.1017G>A mutation in exon 2 of the SMPD1 gene, which resulted in a stop codon at codon 339 (designated W339X) in the cat (Fig. 7). The possible effect of the genetic mutation on the ASM expression was evaluated by RT-PCR, immunohistochemistry, and western blot. The mutation site of the SMPD1 mRNA of the cat was much less amplified than that of an unaffected control cat (Fig. 8). Immunohistochemical analysis revealed abundant ASM-positive granules in the neuronal cytoplasm and neuropil in the brain of an unaffected control cat, but only a few ASM-positive granules were found in the present affected cat (Supplemental Figs. S12, S13). Western blot analysis for ASM detected one distinct band with a molecular weight of approximately 60 kDa, which is the molecular weight of ASM, in the brain samples from an unaffected control cat and the present affected cat. No significant changes were observed between the present cat and an unaffected cat by western blot analysis (Supplemental Fig. S14).
In the present case, we confirmed degeneration of neuronal cells and infiltration of macrophages with accumulation of cytoplasmic granules throughout the CNS, PNS, and visceral organs. Ultrastructurally, numerous cytoplasmic concentric bodies were found in the brain and spleen, and stacked membranous materials were found in the liver in addition to the concentric bodies. Furthermore, we detected a nonsense mutation (c.1017G>A) in exon 2 of the SMPD1 gene. Therefore, the present case was diagnosed as feline NPD with a mutation in the SMPD1 gene, a syndrome analogous to human type A NPD.
As far as we know, there have been 6 reported cases of feline type A NPD. 2,4,6,12,13,16 Distribution of the lesions and histological findings in the present case were comparable to those of the previous feline type A NPD cases, indicating that the present case can be included in the same disease category. Previous reports revealed a deficient activity of ASM in the affected cats; however, genetic analysis was not performed. In the present study, we identified decreased ASM reactivity of the brain by immunohistochemistry and a mutation in the feline SMPD1 gene associated with type A NPD.
Infiltration of macrophages has been also detected in visceral organs of human and feline type A and type C NPD. 7,15 The documented cases of feline type A NPD demonstrated infiltration of macrophages in the CNS, lung, liver, and kidney, and degeneration of the CNS neuronal cells and various epithelial cells. 2,6,13,16 In the present case, we additionally found infiltration of macrophages in the alimentary tract, pancreas, and adrenal glands, and degeneration of neuronal cells throughout the PNS. Human type A NPD patients often exhibit these PNS pathology. 7 In human cases, NPD is clinically diagnosed by detecting either a deficient enzyme activity or a genetic mutation. 7 However, in most feline type A and type C NPD cases, affected cats die before the age of 20 months, 2,6,13,15 –17 and rapid diagnosis is required. Therefore, detection of foamy cells in visceral or PNS tissues may be useful for rapid and simple diagnosis of feline type A and type C NPD.
Some morphological features of the CNS lesions in NPD, including degenerated neurons and macrophages, are similar to those found in other feline lysosomal diseases. 5,7,11 In the present case, cytoplasmic granules in neuronal cells were negative for LFB and PAS and were membranous concentric structures in electron microscopy (EM), while those in macrophages were positive for LFB, but negative for PAS. In feline type C NPD, cytoplasmic granules in neuronal cells were also negative for LFB and PAS 17 and were membranous concentric structures in EM. 1 This histochemical property has been described for distinguishing NPD from other lysosomal diseases in human. 7 In feline Krabbe’s disease (globoid cell leukodystrophy), accumulated granules in the cytoplasm of macrophages (globoid cell) are positive for PAS and were fine tubular structures in EM. 11 In feline GM2-gangliosidosis, accumulated granules of degenerated neuronal cells and macrophages are positive for LFB and PAS and had membranous concentric structures in EM. 5 In feline neuronal ceroid lipofuscinosis, accumulated granules of degenerated neuronal cells are positive for LFB and PAS, exhibit green-yellow autofluorescence by fluorescence microscopy, and are oval to round highly electron-dense structures in EM. 3 Therefore, these histochemical properties may be useful to differentiate feline lysosomal storage diseases.
Biochemical and genetic analysis identified the homozygous nonsense mutation (c.1017G>A) in exon 2 of the SMPD1 gene and a decrease of the ASM immunoreactivity by immunohistochemistry in the present case. In general, nonsense mutations lead to the generation of nonfunctional truncated proteins or to the generation of mRNA species that are rapidly eliminated by nonsense-mediated decay (NMD), a pathway that degrades transcripts with a premature termination codon (PTC). 9,18 Target transcripts during NMD are degraded near the PTC by an endonuclease such as SMG6 in the splicing process. 9 In this case, the SMPD1 mRNA including the mutation site was not amplified by RT-PCR, though upstream and downstream of the mutation site were amplified normally. Therefore, we speculated that the SMPD1 mRNA might be degraded near the PTC by the NMD. The ASM protein consists of an N-terminal saposin domain and a C-terminal catalytic domain. 8 In this case, the nonsense mutation occurred at the central region of the catalytic domain. In human cases, 13 nonsense mutations of the SMPD1 gene have been identified, but the c.1017G>A (p.W339X) mutation observed in the present feline case has not been reported. 18 In humans, 3 SMPD1 nonsense mutations (p.W32X, p.E260X, and p.L263X) have been examined, and all of them lead to a severe loss of ASM activity (less than 5% of wild type). 18 Taken together, we speculate that that case represents severe reduction of ASM expression and enzyme activity as the cause of NPD in a cat with a mutation in the SMPD1 gene.
Supplemental Material
Supplemental Material, Combined_supplemental_materials-Takaichi_et_al - Feline Niemann-Pick Disease With a Novel Mutation of SMPD1 Gene
Supplemental Material, Combined_supplemental_materials-Takaichi_et_al for Feline Niemann-Pick Disease With a Novel Mutation of SMPD1 Gene by Yuta Takaichi, James K. Chambers, Mun Keong Kok, Hiroki Uchiyama, Makoto Haritani, Daisuke Hasegawa, Hiroyuki Nakayama and Kazuyuki Uchida in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by a grant-in-aid from Japanese Society for the Promotion of Science (18H02338, 19J22779).
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.