
Abstract
A glycogen storage disease affecting primarily the skeletal muscle and, to a lesser degree, the cardiac muscle, spinal cord, and brain was diagnosed in a 10-year-old neutered Abyssinian cat with a 4-year history of paresis progressing to acute paralysis. Microscopically, these tissues contained inclusions that were pale basophilic in hematoxylin and eosin-stained slides, diastase resistant, periodic acid-Schiff positive, and blue-to-almost black with iodine stain. By transmission electron microscopy, the inclusions consisted of cytosolic, usually sharply demarcated, nonmembrane-bound deposits of finely granular and filamentous material. on the basis of the structural and histochemical staining characteristics, the inclusions were believed to be aggregates of abnormally stored, unbranched glycogen. A defect in glucose metabolism is suspected to be the underlying pathologic process, but an exact cause remains elusive.
Keywords
Idiopathic polysaccharide storage disease affecting the striated skeletal and cardiac muscles as well as the central nervous system in an adult Abyssinian cat is described. Hitherto in the feline species, glycogen storage disease affecting multiple tissues has been reported only in Norwegian forest cats as a lethal inherited metabolic disease due to a deficiency of glycogen branching enzyme (glycogen storage disease type IV). 5
A 10-year-old neutered Abyssinian cat was originally presented to the Veterinary Teaching Hospital at Purdue University in 1999 with a 2-week history of hind limb paresis. Neurologic examination was otherwise normal. The presumptive diagnosis of myasthenia gravis was made on the basis of the clinical signs, but a serum acetylcholine receptor binding antibody test had a low borderline titer (0.29 nmol/liter; normal reference range for cats, <0.3 nmol/liter), and the results obtained with electromyography were equivocal. The animal was temporarily lost for further workup and was presented to the Veterinary Teaching Hospital for euthanasia approximately 4 years later due to acute posterior paralysis and renal failure. Clinical pathologic alterations obtained at this time included moderate leukocytosis (37.9 × 103/μl), mature neutrophilia (36.0 × 103/μl), and mild lymphopenia (1.4 × 103/μl), which were interpreted as an established inflammation possibly with superimposed stress, mild micro-cytic hypochromic anemia (hematocrit, 24.6%; mean corpuscular volume, 35.4 fl; mean corpuscular hemogloblin, 12.5 pg), probably due to iron deficiency, apparently controlled diabetes mellitus (glucose, 117 mg/dl), a suppressed serum thyroxine level (T4, 0.36 μg/dl), and renal insufficiency (serum urea nitrogen, 211.2 mg/dl; creatinine, 6.4 mg/dl; phosphorus, 17.2 mg/dl).
A complete postmortem examination was performed. The main findings consisted of primary hepatic neoplasm, nephrolithiasis, and bilateral renal crest necrosis. No gross abnormalities were observed in the musculoskeletal system.
Specimens of multiple tissues, including caudal thigh muscle, the entire brain, and multiple segments of the spinal cord, were fixed in neutral-buffered formaldehyde (10% formalin) and were then paraffin embedded, sectioned, and stained with hematoxylin and eosin (HE), periodic acid–Schiff (PAS) with and without diastase digestion, and 0.15% aqueous iodine potassium iodide as previously described. 11 Specimens of the formalin-fixed skeletal muscles of the caudal thigh and spinal cord were postfixed in glutaraldehyde (phosphate buffer), followed by osmium-ferrocyanide, and were then stained with uranium and lead and examined with a transmission electron microscope.
Microscopically, approximately 35% of the skeletal myo-fibers contained many 1- to 7-μm-diameter round-to-ovoid inclusions, which were more numerous in the center of the affected fibers than on their periphery (Figs. 1, 2). The inclusions stained pale basophilic with HE and red-purple with PAS, were diastase resistant and blue-to-almost black with the iodine stain, and were dark blue in methylene blue-azure II-stained semithin sections. Several myofibers were shrunken (atrophic) or enlarged (hypertrophic), whereas others were degenerated and characterized by swollen, eosinophilic, homogeneous or fragmented sarcoplasm and loss of cross-striation. Internalized nuclei were common. Scattered infiltrating macrophages contained material with a staining affinity similar to that of the muscle inclusions. The interstitial adipose tissue was slightly increased. Cardiomyocytes had rare intrasarcoplasmic amphophilic-to-basophilic storage material with tinctorial characteristics similar to the material described for the skeletal muscle.
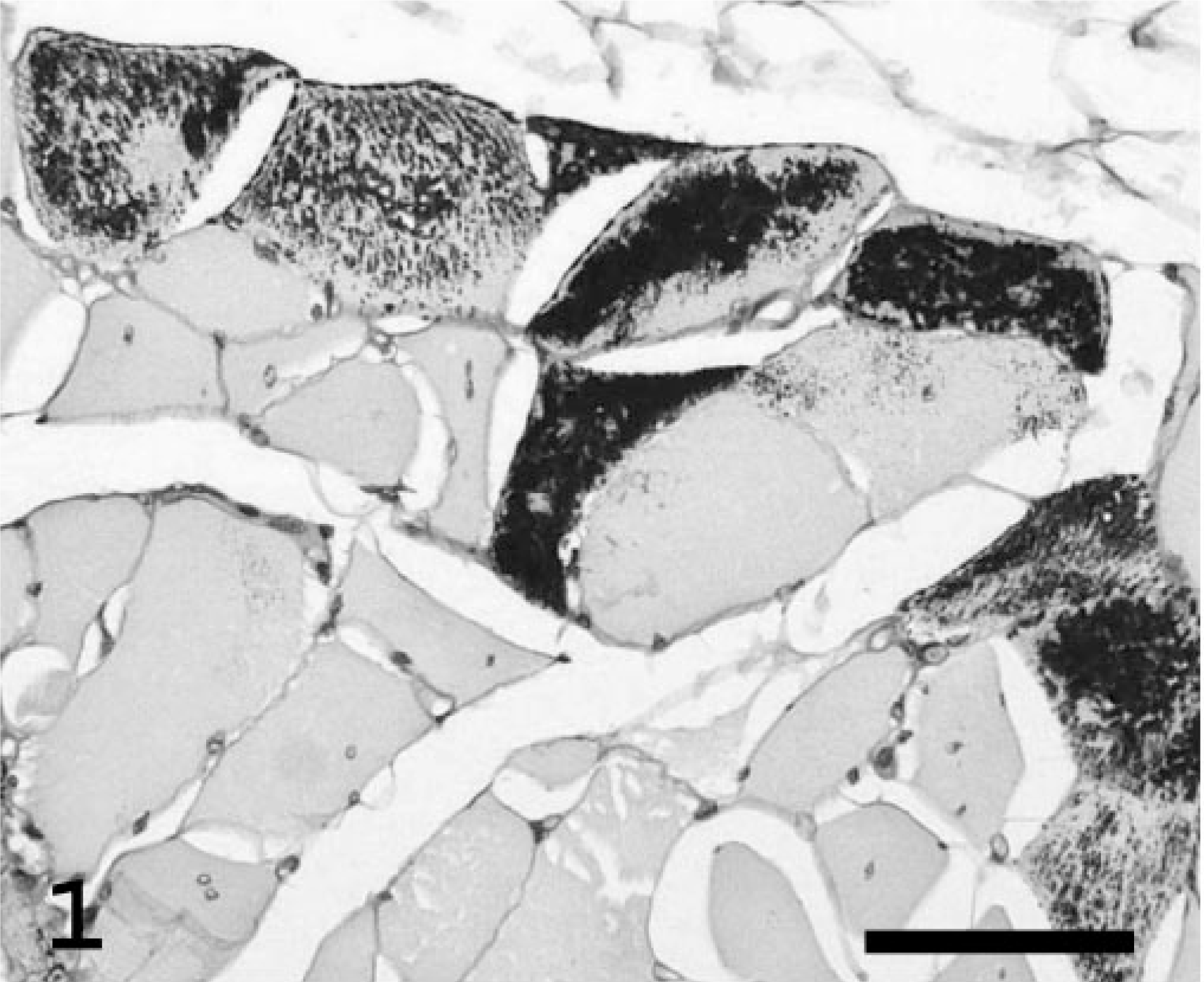
Skeletal muscle, thigh; Abyssinian cat. Transverse section of muscle with many PAS-positive, diastase-resistant intrasarcoplasmic inclusions (dark granules) in several muscle fibers. Note the marked variation in fiber diameter and the presence of internalized nuclei. PAS with diastase. Bar = 50 μm.
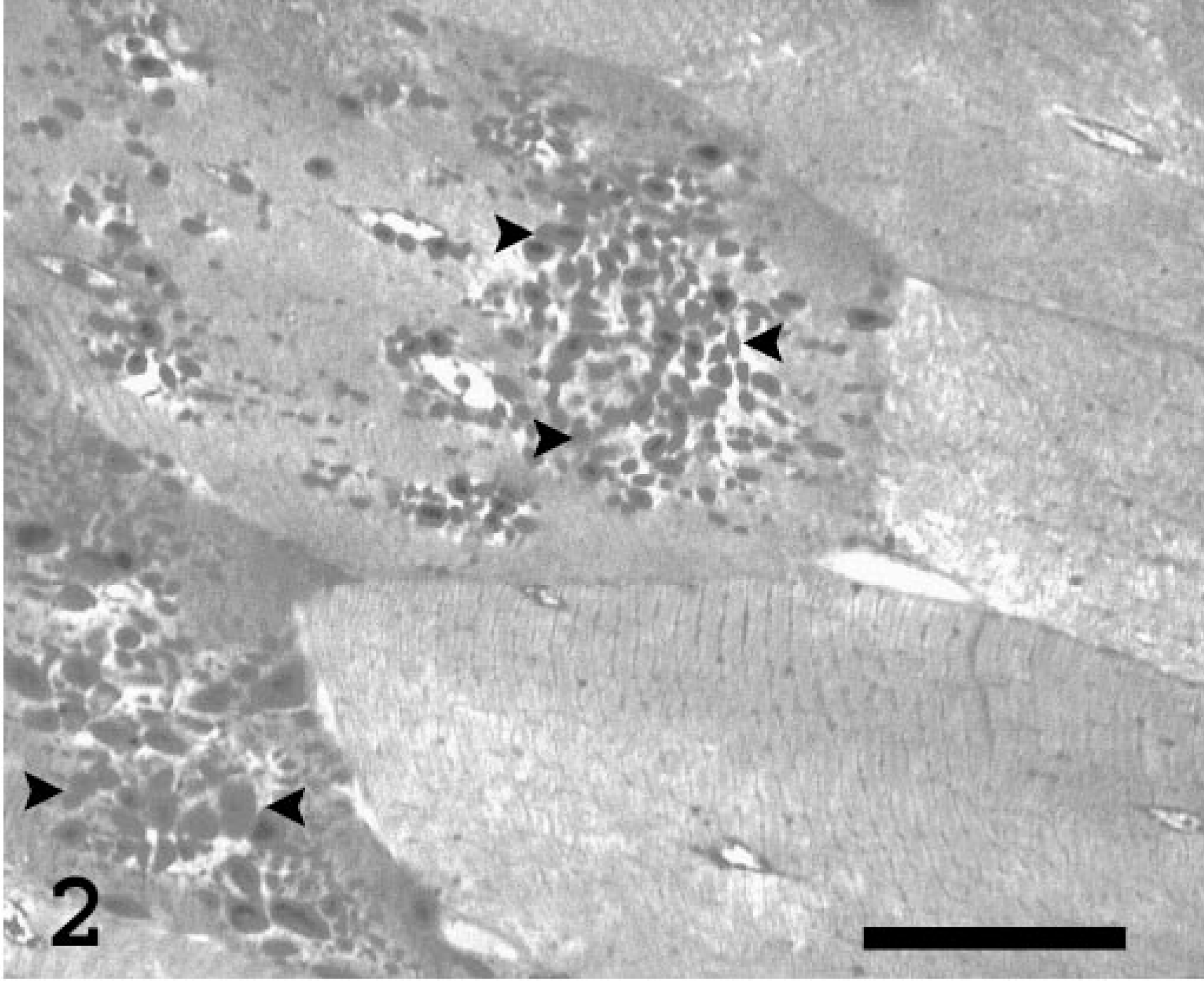
Skeletal muscle, thigh; Abyssinian cat. Higher magnification of the polysaccharide inclusions (arrowheads) in the sarcoplasm of two myofibers. Methylene blue-azure II. Plastic embedded semithin section. Bar = 25 μm.
Spherical, 3- to 10-μm, pale basophilic inclusion bodies were also scattered throughout the central nervous system, predominantly in nerve processes in the neuropil of cerebellar and brain stem nuclei (such as in the lateral cuneate nuclei, nuclei of spinal tract of trigeminal nerves, hypoglossal nuclei, and parasympathetic nuclei of the vagus nerve) and in the gray matter of the entire spinal cord, affecting the dorsal and ventral horns equally (Fig. 3). The abnormal storage product was readily detectable by PAS and was resistant to diastase digestion. Additional changes consisted of multifocal mild axonal swelling and dilated myelin sheaths with occasional macrophages. The sections of sciatic nerve were unremarkable. No PAS-positive inclusions were noted in the liver, smooth muscle of the stomach and intestine, cells of the splenic and mesenteric nodal reticuloendothelial system, kidney, oral mucocutaneous junction, or sciatic and intramuscular nerves.
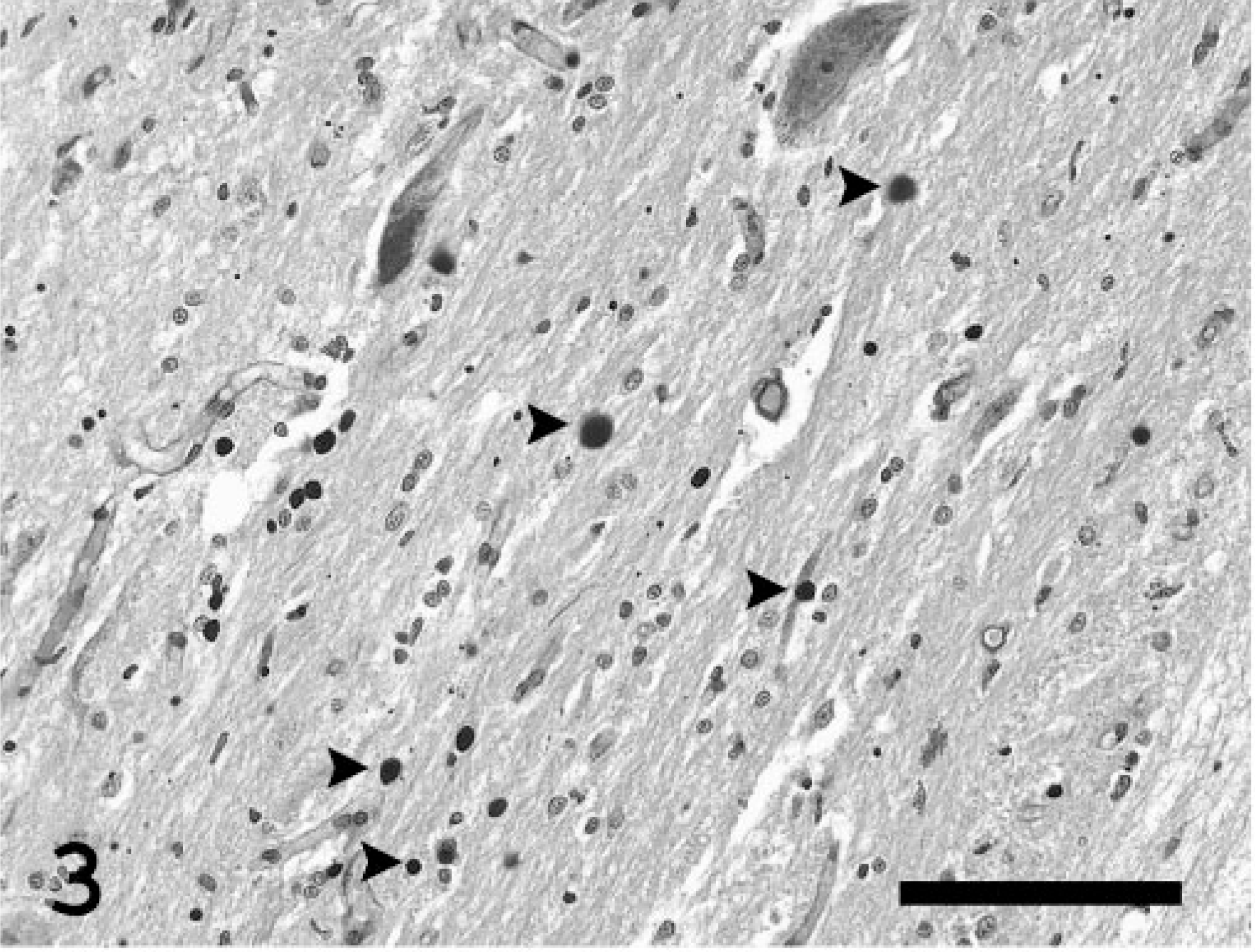
Spinal cord; Abyssinian cat. Numerous spherical polysaccharide inclusions (arrowheads) are scattered throughout the gray matter. PAS with diastase. Bar = 75 μm.
Other histopathologic findings in this cat included cholan-giocarcinoma, cystitis, pyelonephritis, renal crest necrosis, and amyloidosis of variable severity affecting 80–85% of the pancreatic islets.
By transmission electron microscopy following a standard processing method, the abnormal deposits in the muscle and neuropil were cytosolic and usually sharply demarcated but not membrane bound (Figs. 4–6). They consisted of a mixture of finely granular and tangled, randomly oriented, filamentous profiles about 6 nm wide. To a variable extent, the inclusions displaced the normal cellular organelles in the sarcoplasm of the affected myofibers as well as in the non-myelinated neural cell processes in the neuropil. Accumulation of normal-appearing glycogen granules was not present.
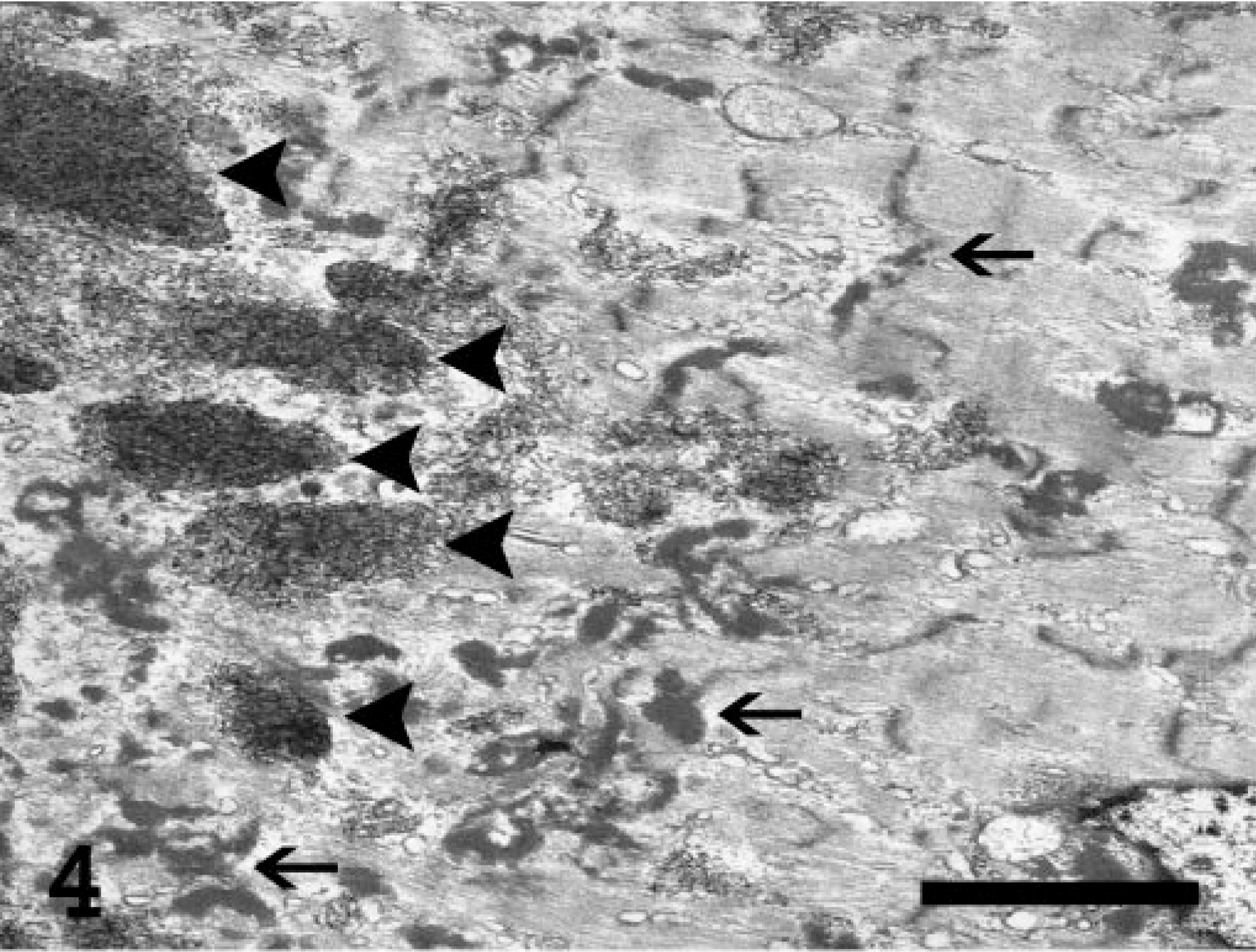
Electron micrograph. Myofiber from the skeletal muscle of the thigh; Abyssinian cat. Several variably sized inclusions (arrowheads) are shown in the sarcoplasm of a myofiber. Degeneration of the affected fiber is indicated by Z-band streaming (arrows) and myofilament disarray. Uranyl acetate–lead citrate stain. Bar = 1.0 μm.
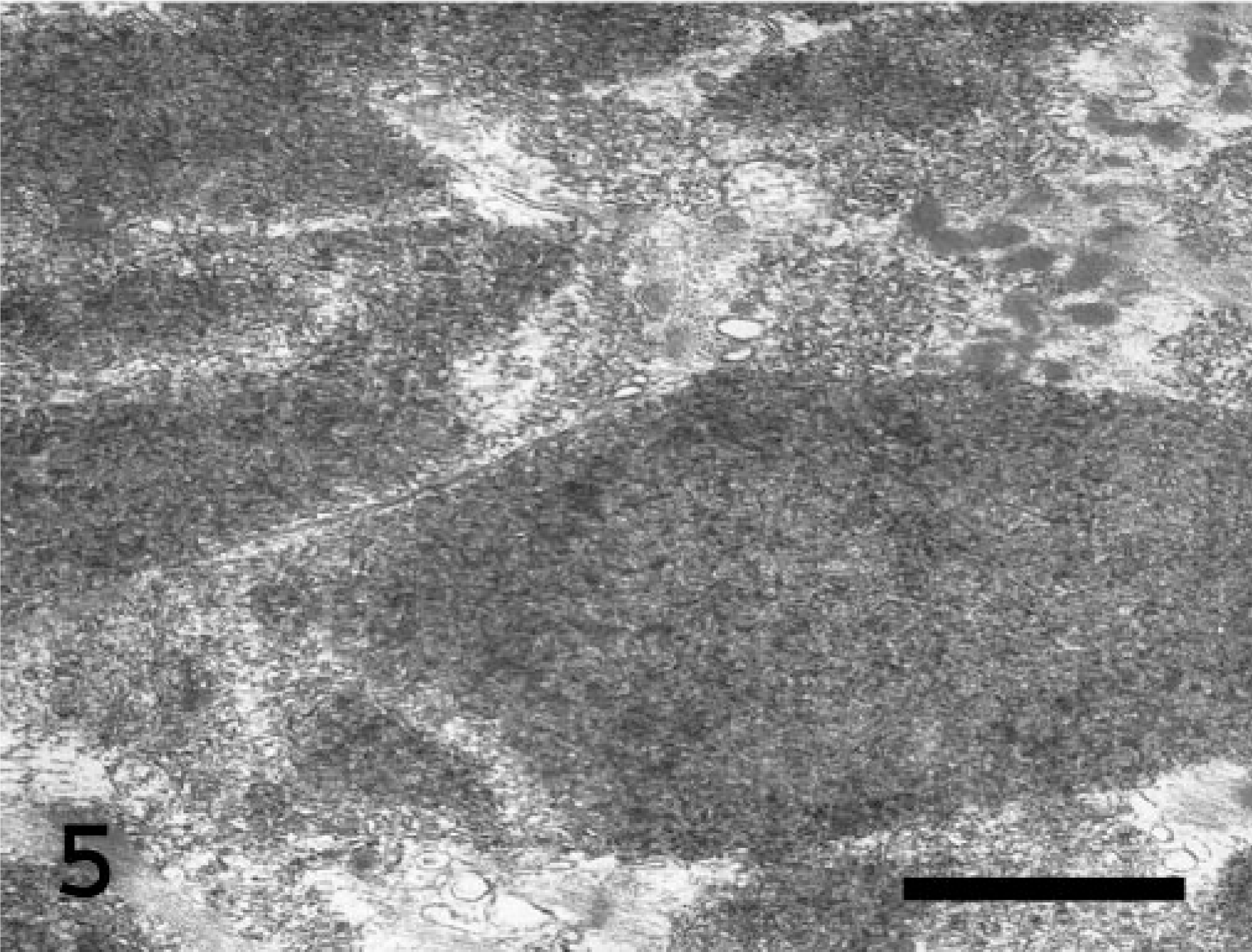
Electron micrograph. Myofiber from the skeletal muscle of the thigh; Abyssinian cat. Nonmembrane-bound, electron-dense, finely granular, and filamentous storage material within the sarcoplasm. Uranyl acetate–lead citrate stain. Bar = 0.16 μm.
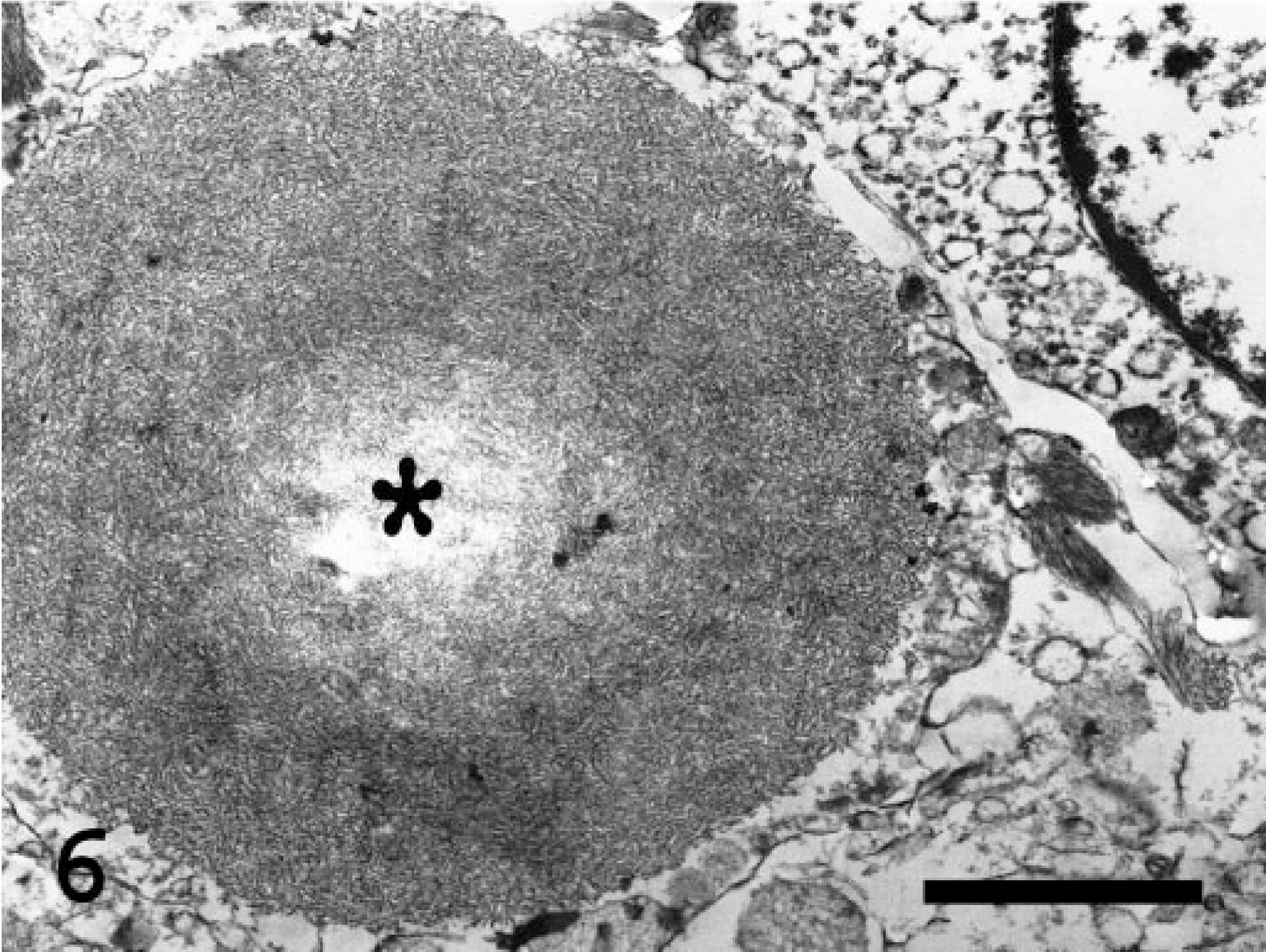
Electron micrograph. Spinal cord; Abyssinian cat. A single round, nonmembrane-bound polysaccaride inclusion (asterisk) nearly completely replaces the cytoplasm in an unidentified cell process. The inclusion is composed of tangled, electron-dense filaments similar to those seen in the skeletal muscle. Uranyl acetate–lead citrate stain. Bar = 0.5 μm.
On the basis of the microscopic and ultrastructural pathologic findings, a presumptive diagnosis of nonlysosomal poly-saccharide storage disease was made. The PAS-positive, diastase-resistant inclusions with blue-to-black staining by iodine and the ultrastructural fibrillar appearance as observed in this cat are consistent with deposits of abnormal glycogen, often referred to as amylopectin, amylopectin-like, corpora amylacea, polyglucosan bodies, Lafora bodies, and complex poly-saccharide. 3 Normal β-glycogen granules are usually present in affected myofibers, and they sometimes surround the fibrillar material that forms the sarcoplasmic inclusions. 3 This feature was not noted in this cat; it may simply be that the normal glycogen was artifactually depleted postmortem or washed out during specimen processing. A defect in glucose metabolism resulting in an accumulation of the abnormal glycogen is, therefore, suspected as the cause of the reported clinical signs in this cat, but an exact cause remains elusive.
In the event of a carbohydrate metabolic defect, a range of polyglucosan bodies can microscopically be found in tissues, including striated muscle and brain. Abnormal accumulations of polysaccharide have also been reported in skeletal muscles adjacent to malignant bone/joint neoplasms in dogs. In the absence of a generalized metabolic disorder, the inclusions were believed to be the result of a local nonspecific metabolic disturbance. 13 In humans, rounded polyglucosan deposits occur in muscle fibers near the myotendinous junction in hypothyroidism. 3 Excessive intrasarcoplasmic accumulations of PAS-positive, amylase-resistant material consistent with abnormally accumulated glycogen have also rarely been reported in hypothyroid animals. 2 The initial workup in this cat included an evaluation of the baseline thyroxine (T4) level, which was on the low borderline concentration (2.3 μg/dl; reference range, 2.4–4.6 μg/dl). Two and a half years later, the animal presented to the referring veterinarian with a marked reduction in the serum T4 level (0.36 μg/dl), but no measurement of thyroid-stimulating hormone was performed to further evaluate the thyroid gland function of this animal. In the absence of the clinical signs typically seen with feline hypothyroidism, namely dermatologic alterations, myxedema of the face, bradycardia, and mild hypothermia, 4 the low serum T4 concentration in this cat was thought to be the result of the suppressive effect of concurrent illness on the hormone concentration (euthyroid sick syndrome), treatment with dexamethasone, or both. For the vast majority of cats, low serum T4 concentrations, even those less than 0.5 μg/dl, are due to the suppression of serum thyroid hormone concentrations in response either to concurrent illness or to the administration of drugs such as glucocorticoids and anti-thyroid hormone drugs. 4 The diagnosis was reinforced by the fact that the thyroid glands of this animal were grossly and histologically unremarkable.
Other disorders that lead to intramyofiber polysaccharide inclusions include canine phosphofructokinase deficiency. In this condition, abnormal glycogen accumulation appears to be a function of age. 7 Clinical signs of hemolytic disease (not seen in the cat of this report) are predominant in dogs affected by this disease, and there are minimal signs of neuromuscular dysfunction. In Lafora's disease in humans, poly-saccharide inclusions accumulate in nervous system tissue as well as, occasionally, in skeletal and cardiac muscles and the liver. 3, 12 Neurologic signs (myoclonus, tremors, and seizures) are typically observed in humans with this disease, 12 whereas they seem to be less common in animals, 6 in which Lafora-like bodies are often reported as incidental changes without apparent neurologic disease. When related to aging, the inclusions in cats tend to occur freely in the neuropil rather than in the cytoplasm of neurons, 8 whereas, when associated with progressive, ultimately fatal neurologic disease, Lafora-like bodies are frequently associated with neuronal perikarya. 6 In this cat, on the basis of light and electron microscopic evaluation, inclusions in the nervous system appeared to be lying either free in the neuropil or within neuronal or astrocytic processes.
An acquired, possibly aging-associated, metabolic defect in glycogen metabolism is thought to be the most likely cause of the myopathy, cardiomyopathy, and encephalomyelopathy in this instance. Less likely would be an inborn error in glucose metabolism leading to a late onset of neuromuscular dysfunction. Iodine stains polyglucans in an aqueous environment in a manner that is roughly proportional to the length of alpha 1,4 linkages uninterrupted by alpha 1,6 linkage (the branch points). 1 Normal glycogen stains with a mahogany brown color. With increasing starchlike characters (longer inner and outer chains without branches), the staining changes from that to mauve, purple, violet, and blue. Because stored abnormal glycogen can be present in dense granules in muscle, the poorly branched glycogen observed in branching enzyme deficiency stains almost black (J. Fyfe, personal communication). Consequently, the blue-to-almost black intracytoplasmic deposits observed in this cat are consistent with unbranched glycogen. The storage material in the muscle and nervous tissue in this cat is, therefore, histochemically and ultrastructurally similar to that in cats with branching enzyme deficiency. Most humans with an adult onset of myopathy, with or without cardiomyopathy and associated with carbohydrate metabolic disorders, have been identified as having idiopathic polysaccharide storage disease. 3 In this condition, the pathologic alterations are reminiscent of brancher deficiency, but the branching enzyme activity is reported as normal. In Norwegian forest cats affected by type IV glycogenosis, the glycogen branching enzyme activity in the liver and muscle was less than 5% and 25–75% of the normal levels in affected animals and normal carriers, respectively. 5 In the cat of our study, no biochemical enzyme assays could be performed because of the lack of fresh samples of muscle, liver, or heparinized blood.
Additional gross and microscopic findings in the urinary tract and the endocrine pancreas in the cat of this study were believed to be the cause of the reported renal failure and diabetes mellitus. Spontaneously or alloxan-diabetic rats and, rarely, human patients with diabetes have been reported with polyglucosan bodies in peripheral nerve axons. 9 In feline diabetes mellitus, peripheral neuropathy is a well-recognized debilitating complication, 10 but no association between diabetes and polyglucosan accumulation has been noted in any tissues of affected cats. In the cat of this study, there was no histologic evidence of neuropathy; the sections of sciatic and intramuscular nerves were unremarkable. Furthermore, the initial workup of the animal for its neuromuscular disorder did not show hyperglycemia indicative of diabetes mellitus. The muscle weakness in this cat was preceded by, and therefore not thought to be a complication of, this metabolic disorder.
In summary, this case report describes the microscopic and ultrastructural pathologic alterations in a single 10-year-old Abyssinian cat with a 4-year history of neuromuscular dysfunction. Complex polysaccharide storage material consistent with abnormal, unbranched glycogen was present in the striated and cardiac muscle as well as in the central nervous system of the animal. This report presents the first case of polysaccharide storage disease affecting multiple tissues in a breed of domestic cats other than the inheritably affected Norwegian Forest cats. Although the deposits are likely the result of a disorder in glucose metabolism, whether the pathologic findings represented the specific product of one particular disease or whether several pathologic processes underlaid its production is not known. A muscle and nerve biopsy should be a routine component of the diagnostic plan for animals suspected of having neuromuscular disease. It is only by means of biopsy that such pathologic processes can be fully understood.
Footnotes
Acknowledgements
We thank Dr. W. B. Morrison (School of Veterinary Medicine, Purdue University, West Lafayette, IN) for referral of this animal, Dr. B. A. Valentine (College of Veterinary Medicine, Oregon State University, Corvallis, OR) for assistance in the histologic evaluation of the muscle alterations, Dr. J. C. Fyfe (Laboratory of Comparative Medical Genetics, Michigan State University, East Lansing, MI) for assistance in the use and interpretation of the iodine stain, and Drs. J. F. VanVleet and J. A. Ramos-Vara (School of Veterinary Medicine, Purdue University, West Lafayette, IN) for helpful discussion and critical review of this manuscript.