
Abstract
Three young domestic shorthair cats were presented for necropsy with similar histories of slowly progressive visual dysfunction and neurologic deficits. Macroscopic examination of each cat revealed cerebral and cerebellar atrophy, dilated lateral ventricles, and slight brown discoloration of the gray matter. Histologically, there was bilateral loss of neurons within the limbic, motor, somatosensory, visual, and, to a lesser extent, vestibular systems with extensive astrogliosis in the affected regions of all 3 cases. Many remaining neurons and glial cells throughout the entire central nervous system were distended by pale yellow to eosinophilic, autofluorescent cytoplasmic inclusions with ultrastructural appearances typical of neuronal ceroid-lipofuscinoses (NCLs). Differences in clinical presentation and neurological lesions suggest that the 3 cats may have had different variants of NCL. Molecular genetic characterization in the 1 cat from which DNA was available did not reveal any plausible disease-causing mutations of the CLN1 (PPT1), CLN3, CLN5, CLN8, and CLN10 (CTSD) genes. Further investigations will be required to identify the mutations responsible for NCLs in cats.
The neuronal ceroid-lipofuscinoses (NCLs) are recessively inherited neurodegenerative lysosomal storage diseases characterized by the accumulation of autofluorescent lipopigments within the central nervous system (CNS) and peripheral tissues. 11 NCLs have been documented in a number of domestic animal species, predominantly in dogs,1,2,7,13–15,19,20,22,24,28 sheep, 8,12,25,29 cats, 4,9,10,18,21,30 and cattle. 12,26 Affected animals typically present with a clinical history of progressive cognitive decline, motor dysfunction, vision deficits, and seizures. Neurological decline ultimately results in death or the necessity for euthanasia. Among both people and dogs, differences in clinical presentation, nervous system pathology, and the ultrastructural appearance of the lysosomal storage material are almost always associated with differences in the underlying mutations. 1 –3,7,14,19,26,28
More than 360 mutations in at least 14 different genes coding for a variety of proteins, including lysosomal enzymes and membrane proteins associated with lysosomes, endoplasmic reticulum, endoplasmic reticulum–Golgi intermediate compartment membranes, and synaptic vesicles, underlie the human forms of NCL. 17 To date, in the veterinary literature, characterization of spontaneous causative mutations of NCL candidate genes has largely been limited to dogs (ARSG, 1 ATP13A2, 7 PPT1 (CLN1), 28 TPP1 (CLN2), 13 CLN5, 19 CLN6, 14 CLN8, 15 and CTSD (CLN10) 2 ) and sheep (CLN5, 8 CLN6, 25 and CTSD (CLN10) 29 ) but have also been reported in cattle (CLN5). 26 In dogs, the detection of causative NCL candidate genes has been restricted to purebreds, including American Staffordshire Terriers (ARSG), 1 American Bulldogs (CTSD), 2 Tibetan Terriers (ATP13A2), 7 Dachshunds (TPP1 and PPT1), 13,28 an Australian Shepherd (CLN6), 14 English Setters (CLN8), 15 and Border Collies (CLN5). 19 Similarly, purebred sheep such as Borderdales (CLN5), 8 South Hampshires (CLN6), 25 and White Swedish Landraces (CTSD) 29 comprise the majority of reports of ovine NCL. Analogous to the human forms of NCL, in canine and ovine NCL, mutations primarily occur in genes that code for various lysosomal enzymes (ARSG, ATP13A2, CLN5, CTSD, PPT1, TPP1), endoplasmic reticulum proteins (CLN6), and endoplasmic reticulum–Golgi intermediate compartment membranes (canine CLN8). NCL has also been reported in cats, but only limited pathological and molecular genetic analysis has been performed and no causative mutations identified thus far. 9 This report documents 3 cases of suspected feline NCL with sequence analysis of NCL candidate genes in 1 cat.
Clinical Histories
Three cats were submitted to the University of Minnesota Veterinary Diagnostic Laboratory (St Paul, MN) for postmortem examination between 2008 and 2011 with a similar signalment and a history of chronic progressive neurological clinical signs. The cats were all male neutered domestic shorthairs that initially presented with clinical signs at the age of 6 months (cat No. 2), 1.5 years (cat No. 3), and between 1 and 2 years (cat No. 1; age estimated). The progression of clinical disease from onset to euthanasia was approximately 1 year in cat Nos. 2 and 3; however, this could not be determined for cat No. 1 (stray animal). Neurological clinical signs in all 3 cats were largely characterized by disorientation, partial (facial) and generalized seizures, auditory and/or tactile hyperesthesia, and variable visual deficits (blindness, absent menace reflex). Cat No. 2 also exhibited maniacal running, compulsive pacing, sporadic myoclonus, and cataplexy. Due to the severity of clinical signs and quality-of-life issues, humane euthanasia was elected in all 3 cases. Pedigree analysis was not available for any of the cats.
Gross and Microscopic Findings
At necropsy, macroscopic examination of the brain of all 3 cats revealed mild to moderate, diffuse, symmetrical cerebral and cerebellar atrophy. The cortical gyri and cerebellar folia were extensively flattened and narrow, sulci and fissures were mild to moderately widened, the lateral ventricles were mild to moderately dilated, and there was dull brown discoloration of the entire cerebral and cerebellar cortices. Examination of other organs and tissues in these cats did not reveal any significant macroscopic lesions.
Samples were collected from various organs and tissues, including brain, spinal cord, eyes, heart, liver, lungs, kidneys, gastrointestinal tract, adrenal gland, thyroid gland, and skeletal muscle; fixed in 10% neutral buffered formalin; routinely processed (ie, embedded in paraffin wax, sectioned at 5 μm); and stained with hematoxylin and eosin (HE). Sections of brain were also stained with periodic acid–Schiff (PAS), Luxol fast blue (LFB), and Ziehl-Neelsen acid-fast blue (AFB) stains. Selected portions of formalin-fixed brain were also postfixed in 1% osmium tetroxide and embedded in Embed (Electron Microscopy Sciences, Hatfield, PA), from which 80-nm ultrathin sections were stained with uranyl acetate and lead citrate (Electron Microscopy Sciences) for ultrastructural examination. Immunohistochemical staining was performed on brain sections from all 3 cats and an unaffected 2-year-old domestic shorthair cat (aged- and size-matched control) using a peroxidase-based polymer system (Envision TM-HRP; Dako, Carpinteria, CA). Primary antibodies included synaptophysin (1:50, monoclonal, clone SY38; Dako), glial fibrillary acidic protein (GFAP; 1:600, polyclonal; Dako), glutamic acid decarboxylase 65+67 (GAD; 1:200, polyclonal; Millipore, Temecula, CA), and γ-aminobutyric acid (GABA) plasma membrane transporter 1 (GAT-1; 1:100, polyclonal; Abcam, Cambridge, MA). Immunopositive reactions were visualized with the chromogen, 3-amino-9-ethylcarbazole, and sections were counterstained with Mayer’s hematoxylin. Unstained sections of brain and liver from all cases were evaluated with a fluorescence microscope using ultraviolet light excitation (DAPI filter; excitation, 320–390 nm; emission, 430–490 nm), blue light excitation (FITC filter; excitation, 455–500 nm; emission, 500–570 nm), and green light excitation (TRITC filter; excitation, 505–560 nm; emission, 575–655 nm).
In all cases, microscopic examination of HE sections revealed a moderate to severe loss of neurons within the cerebrocortical laminae of the frontal (motor system) and parietal (somatosensory system) cortices and neurons of the parahippocampal, cingulate, and subcallosal gyri (limbic lobe; limbic system). There was mild to moderate neuronal loss within the hippocampus (limbic system) and moderate loss within the lateral geniculate nucleus (visual system). Within the cerebellum, there was minimal to mild spongy degeneration of the granular cell layer and mild diminution of Purkinje, glomerular, and granule cells. Throughout the affected regions of the cerebrum and cerebellum and in some random segments of the brainstem and the ventral horn of the spinal cord, neurons and glial cells were moderate to severely distended by 1- to 6-μm diameter, pale yellow or pale eosinophilic, botryose, glassy, intracytoplasmic vacuoles, which in some cells were associated with marginalization of nuclei (Fig. 1a). The content of these vacuoles was intensely positive by LFB stain (Fig. 1b), mild to moderately positive by PAS reaction (Fig. 1c), and negative by AFB stain, and it exhibited autofluorescence using a fluorescence microscope (Fig. 1d). The most robust autofluorescence was observed using the FITC excitation filter (excitation, 455–500 nm) and was associated with the emission of green fluorescence. Autofluorescence was also observed with the DAPI and TRITC excitation filters, but emission levels were subjectively below that observed with the FITC excitation filter. There were markedly increased numbers of astroglia unevenly distributed throughout both the cerebral and cerebellar gray and white matter, especially within the cortical laminae affected by neuronal cell loss.
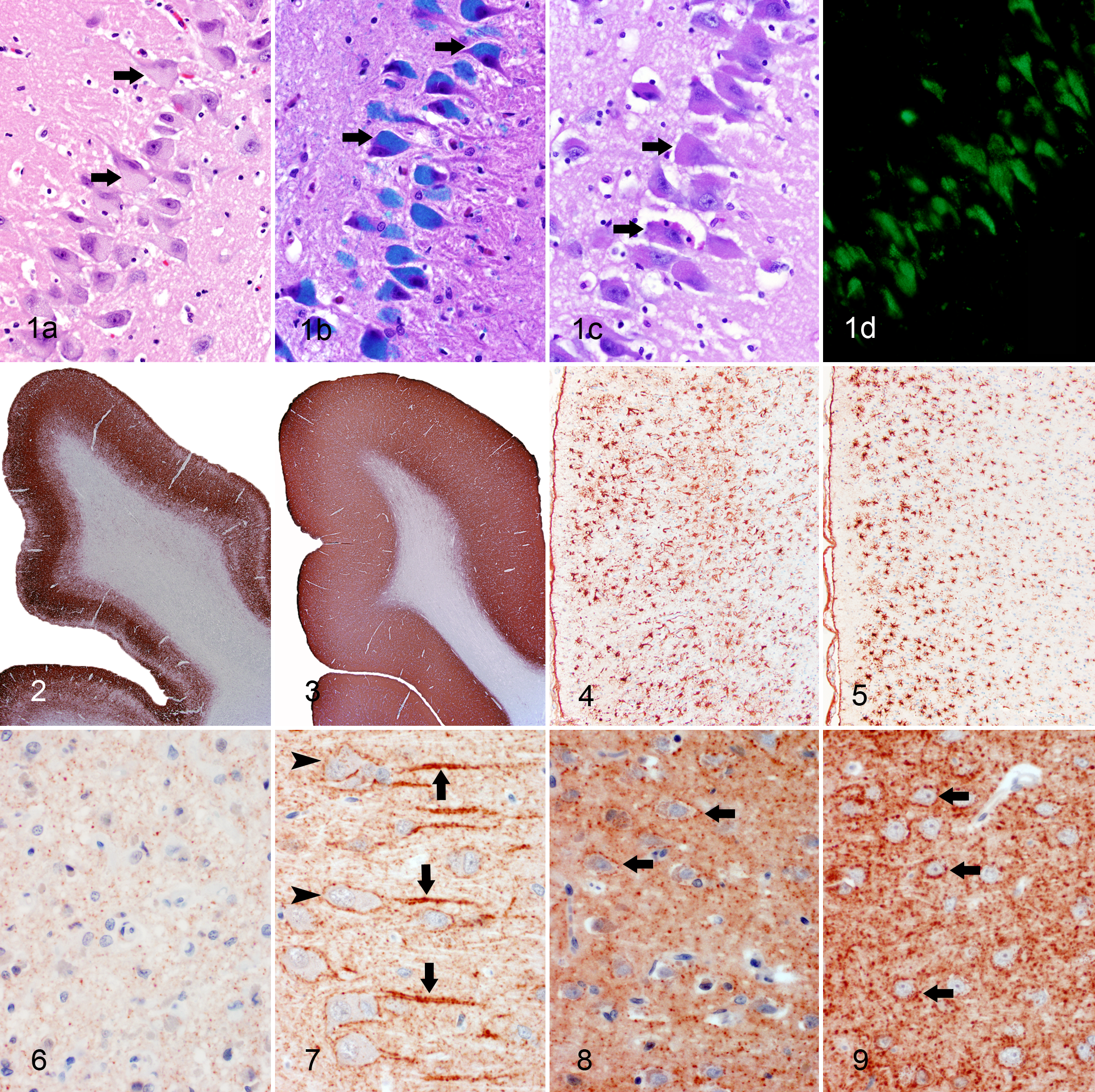
Hippocampus, cornu ammonis 2 (CA2) region; cat No. 3. Pyramidal neurons (arrows) contain abundant pale yellow, pale eosinophilic, or acidophilic intracytoplasmic storage material on hematoxylin and eosin stain (a), which is intensely Luxol fast blue positive (b), is mildly periodic acid–Schiff positive (c), and exhibits green-yellow autofluorescence by fluorescence microscopy (d). Fluorescence microscope; FITC filter (excitation, 455–500 nm; emission, 500–570 nm).
Relative to cat Nos. 1 and 2, cat No. 3 exhibited the greatest overall CNS neuronal/cell loss and gliosis when compared across most functional/anatomic systems. Based on the distribution and level of storage material accumulation, it was difficult to differentiate the 3 cases. Bilaterally, in cats Nos. 1 and 2, there was extensive disorganization of the inner and outer retinal nuclear layers with diminution of the constituent cells and rarefaction of the plexiform layers. There was a paucity of ganglion cells, the remaining of which occasionally contained intracytoplasmic storage vacuoles with a similar appearance to the affected neurons within the brain and spinal cord. The retina of the 1 eye examined histologically in cat No. 3 appeared essentially normal. Although all 3 cases shared a similar distribution and pattern of lesions, the intensity and severity of the lesions from region to region throughout the CNS varied between the cases. There were no significant histological lesions in other organs and tissues in these cases (including the autonomic and peripheral nervous systems), but a low level of green-yellow autofluorescent material (FITC filter) was observed in the livers of all 3 cats. The relative distribution and severity of microscopic changes for all 3 cases are summarized in Table 1. The severity scoring of microscopic changes in a given location was based on a semi-quantitative scale, relative to a randomly selected age- and breed-matched control. The semi-quantitative scale assessed (1) the estimated quantity of neurons containing storage material, (2) the estimated quantity of neurons lost, and (3) the estimated increase in glial cells. Changes less than one-third were defined as mild, changes between one-third and two-thirds were defined as moderate, and changes greater than two-thirds were defined as severe. Diffusely within the cerebral cortex in all 3 cases, there was a narrowing of the gray matter as subjectively illustrated by synaptophysin immunoreactivity (Fig. 2) relative to the control brain (Fig. 3). The GFAP immunoreactivity throughout the cerebral cortex was more intense and unevenly distributed (Fig. 4) relative to the control brain (Fig. 5). This pattern corresponded with a regional increase in astroglia containing widely sprawling and erratic cell processes. There were significant decreases in both GAT-1 (Fig. 6) and GAD (Fig. 8) immunoreactivity within the cerebral cortex relative to the control brain (Figs. 7 and 9, respectively), presumably secondary to diminished GABAergic neurons. Morphometric quantification was not conducted on the immunohistochemical sections.
Distribution and Severity of Histological Lesions of 3 Domestic Shorthair Cats With Neuronal Ceroid-Lipofuscinosis.a
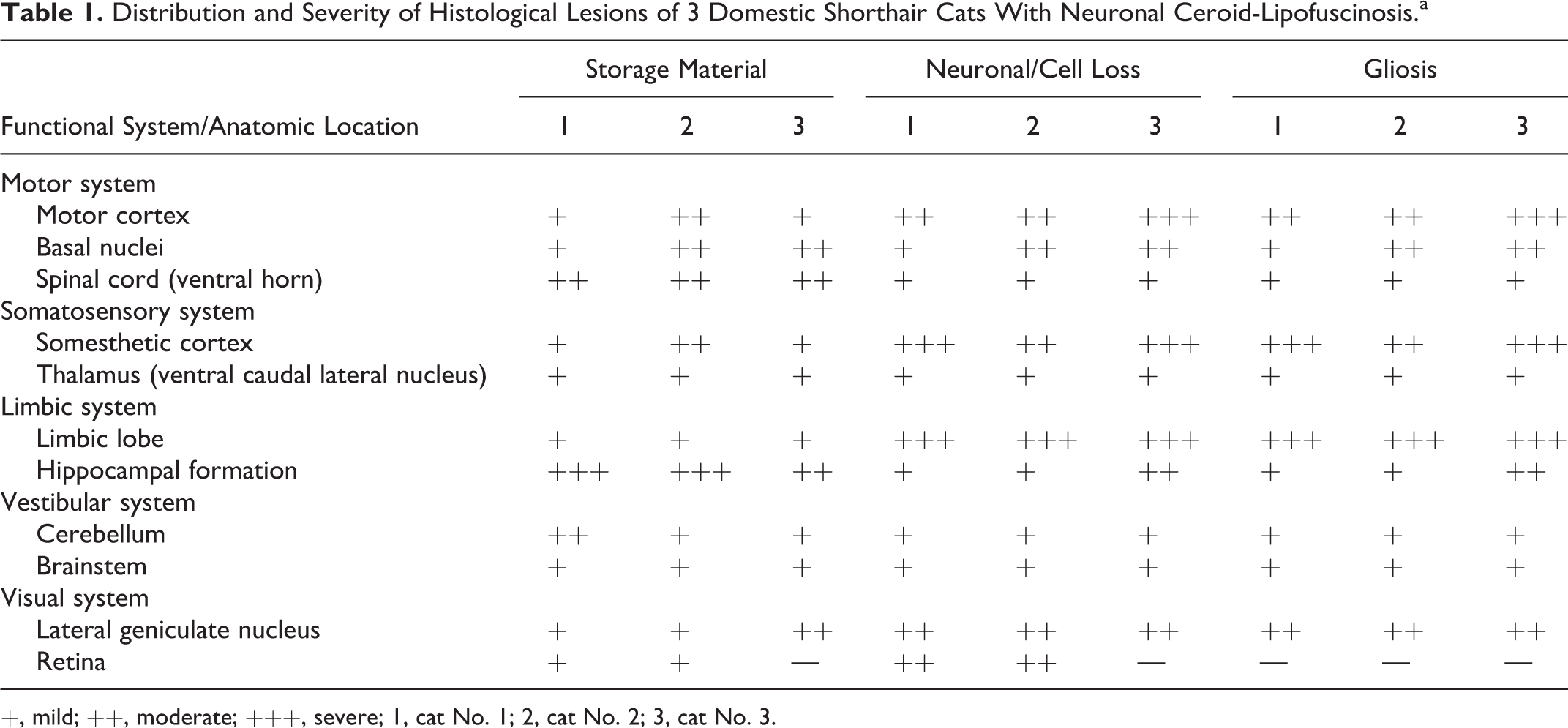
+, mild; ++, moderate; +++, severe; 1, cat No. 1; 2, cat No. 2; 3, cat No. 3.
In cat No. 1, ultrastructural examination of cerebral and cerebellar neurons indicated that the intracytoplasmic storage bodies appreciated on histological analysis consisted of membrane-delimited structures containing a variable and mixed accumulation of material (Fig. 10) composed of 1 or more of the following: (1) granular osmophilic deposits (GROD; Fig. 11)—variably sized, oval to round highly electron-dense deposits; (2) curvilinear profiles (CV; Fig. 12)—short, curved, thin, lamellar stacks alternating between low and high electron densities; (3) fingerprint profiles (FP; Fig. 13)—tightly packed concentric membranous stacks; and (4) rectilinear profiles (RL; Fig. 14)—stacked, linear membranous bodies. Comparatively, in cat Nos. 2 and 3, these membrane-delimited structures contained variable amounts of predominantly FP and RL material. Organs other than the brain were not examined ultrastructurally in these cases.
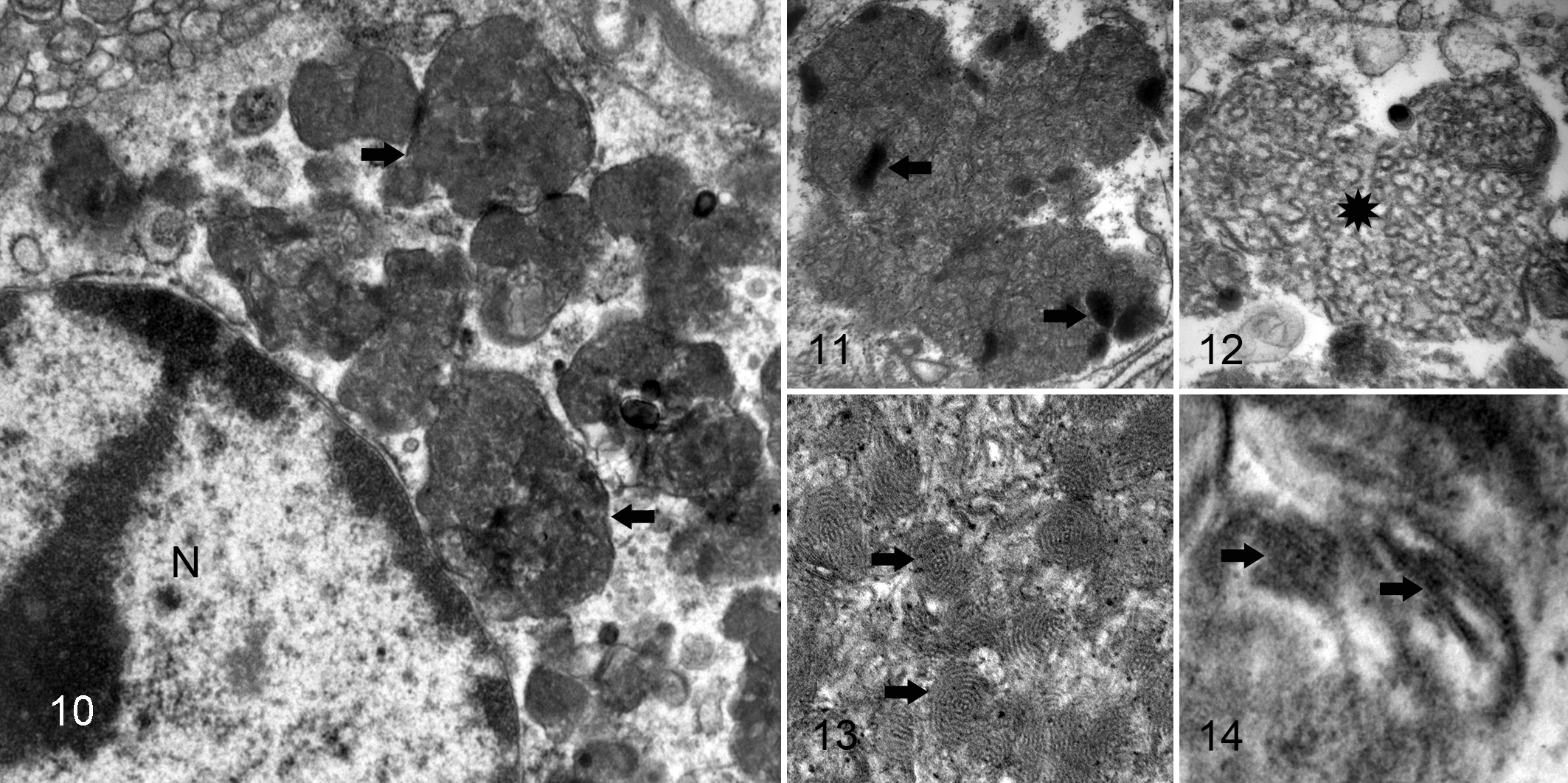
Hippocampus; cat No. 1. A low-power magnification of a pyramidal neuron containing a conglomerate of intracytoplasmic, electron-dense, membrane-bound storage material (arrows). Nucleus (N). Electron microscopy; uranyl acetate and lead citrate.
NCL Candidate Gene Mutational Analysis
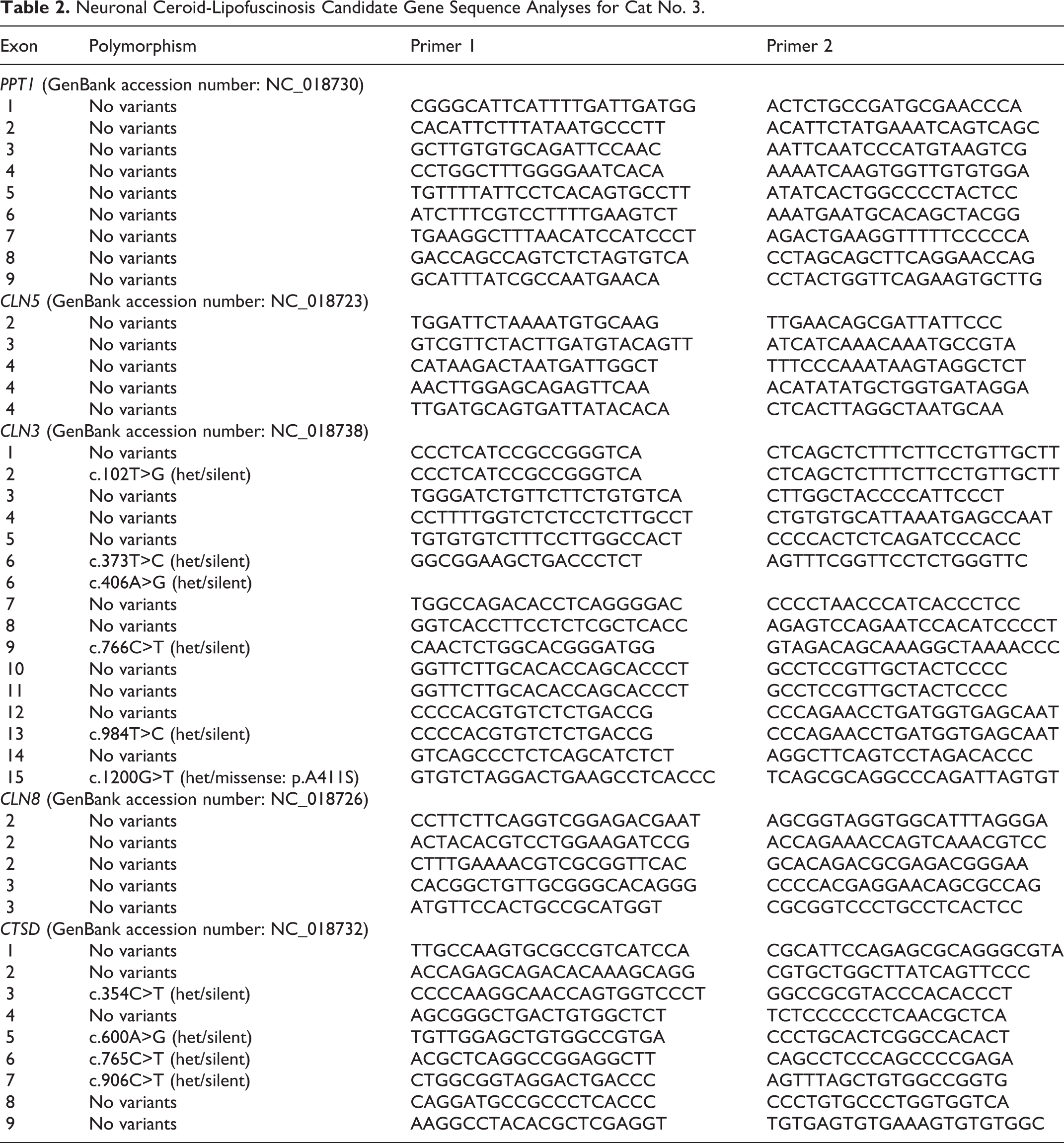
Analyses were performed to assess whether a mutation in one among several genes was associated with the disease in cat No. 3, the only one of the 3 cases from which unfixed tissue suitable for DNA isolation was available. Genomic DNA was isolated from a frozen liver sample from this cat using established and described techniques. 15 Using this DNA as a template, exons and flanking portions of the introns were amplified from selected candidate genes, PPT1 (CLN1), CLN3, CLN5, CLN8, and CLN10 (CTSD), using polymerase chain reaction (PCR) primers designed with Oligo 7 software (Molecular Biology Insights, Cascade, CO) and based on the cat genome reference sequence (www.ncbi.nlm.nih.gov/genome/78). 27 Previously described procedures were used for amplicon purification and automated Sanger sequencing. 31 The sequences were obtained in both directions and individually inspected after alignment to the corresponding reference sequences with Sequencer software (Gene Codes, Ann Arbor, MI). Of the candidate genes evaluated, a number of sequence variants were identified compared with the cat reference sequence (Table 2), but none appeared likely to be a disease-causing mutation. Only 1 of the sequence variants predicted a change in the amino acid sequence of the encoded protein. This sequence variant, in exon 15 of CLN3, predicted an alanine to serine alteration at amino acid 411. However, this mutation was present in only 1 allele of CLN3; the nucleotide on the other allele at this locus was the same as that in the reference sequence. A cross-species comparison of CLN3 sequences in the NCBI database indicates that amino acid 411 is not conserved. All other sequence variants identified in the candidate genes were silent.
Neuronal Ceroid-Lipofuscinosis Candidate Gene Sequence Analyses for Cat No. 3.
Discussion
Consistent with the diagnosis of NCL based on clinical signs, pathological examination of these cats revealed a severe widespread degenerative encephalomyelopathy accompanied by intracellular accumulation of autofluorescent storage material. Comparable with previous reports of feline NCL, these cases were characterized by progressive neurological deficits and vision loss arising at a relatively young age associated with widespread CNS neuronal loss and intraneuronal and glial accumulation of storage material containing LFB-positive, PAS-positive, and autofluorescent lipopigment. 4,9,10,18,21,30 From a diagnostic perspective, these features may not differentiate NCL from other lysosomal storage diseases (eg, gangliosidoses, mannosidoses), but the characteristic ultrastructural findings in the present cases were more consistent with NCL.
Previous case reports and studies in several domesticated species indicate that NCL, as in many of the human forms of the disease, is associated with regional and time- and cell-specific changes in the CNS. 6,23,25,30 Although there may be some inter- and intraspecies variation in the distribution of lesions at the time of death/euthanasia, there is most often a loss of neurons within the cerebral cortex and, to a lesser extent, the cerebellar Purkinje and/or granular cell layers. Astrogliosis typically accompanies neuronal loss particularly within the cerebral cortex. Lipopigment accumulation is often most prominent within hippocampal pyramidal cells, cerebellar Purkinje cells, and, to a lesser degree, neurons of the brainstem and the regions of cerebral neuronal loss.1,4,6,10,13 –15,18 –24,28 –30 The primarily green autofluorescence of the lipopigment in the present 3 cats was consistent with previous reports of NCL in cats, a ferret, and a Vietnamese potbelly pig, 4,6,21,23 but the storage material in most dogs with NCL tends to exhibit bright yellow autofluorescence at similar excitation wavelengths. 7,13,14,28 Interspecies diversity of the lipopigment constituents and/or differences in the fluorescence microscopy equipment and/or protocol may explain this apparent disparity. The typical distribution of CNS lesions observed in NCL correlates well with the pathological presentations of the present 3 cases. Notably, neuronal loss was most severe in the high hierarchal centers of the cerebral cortex, limbic lobe, basal nuclei, and thalamus. This also correlates well with the reported clinical signs, which were predominantly associated with a loss of cognition and integration. The visual deficits observed in these cats may be explained by a central loss of neurons within the visual cortex and visual pathways (cat Nos. 1–3) and/or peripherally by retinal atrophy and the accumulation of intracytoplasmic lipopigment within retinal ganglion cells (cat Nos. 1 and 2). Retinal atrophy and pigment accumulation in NCL are commonly observed in the human infantile and juvenile forms 11,17 and some canine 13,14,22,28 and ovine forms, 12 but prior to the current report, retinal lesions have been reported only in 1 feline case. 4
It appears that intraneuronal lipopigment accumulation inversely correlates with neuronal loss in NCL. In the present 3 cases, this premise was supported by changes within the hippocampal pyramidal cells where the most prominent lipopigment accumulation occurred, yet neuronal loss was low. The opposite was evident within the cerebrocortical laminae. This observation supports the notion that neuronal loss in NCL is region and cell specific. Murine and ovine NCL studies have demonstrated a regional and cell-dependent vulnerability to neuronal loss most specifically in the cerebrocortical inhibitory GABAergic interneuron population. 16,25 In parallel with these findings, there was a significant loss of cerebrocortical GABAergic neurons in the present 3 cases, as illustrated by a reduction of GAT-1 and GAD immunoreactivity. GAD catalyzes the conversion of glutamate to GABA in GABAergic neurons. Therefore, with a loss of GABAergic neurons or GABAergic neuronal GAD function, there is presumably a consequential loss of GABA-associated interneuronal inhibition and/or elevated glutamate-associated neuronal excitation. Murine and ovine NCL studies have demonstrated that this loss of cerebrocortical GABAergic neurons chiefly occurs in the later stages of disease when spontaneous seizure activity is greatest. 16,25 In accordance with this finding, the progressive worsening of seizure activity in suspected NCL cases (including the 3 cats presented herein) may be rationally explained by elevated neuronal hyperexcitability associated with the progressive loss of GABAergic interneuronal inhibition and/or an elevation in glutamate-associated neuronal excitation. Nonetheless, other contributory mechanisms of seizure generation cannot be ruled out.
While there were remarkable clinical (ie, age of onset, rate of disease progression and clinical signs) and pathological similarities between all 3 cases, their subtle differences may reflect different underlying etiopathogeneses. Of note were the differences in severity of neuronal cell loss and the ultrastructural appearances of the intracytoplasmic storage material. Considerable CNS neuronal/cell loss and gliosis were observed in cat No. 3 relative to cat Nos. 1 and 2, which exhibited mild to moderate loss. Ultrastructurally, neurons in cat No. 1 exhibited a heterogeneous mix of GROD, RL, FP, and CV material, similar to the late infantile and adult-onset human forms, whereas with cat Nos. 2 and 3, the storage material predominantly consisted of FP and RL, similar to the juvenile form of human NCL (CLN3 gene mutation). 11,17 The human and canine NCLs are also heterogeneous with respect to age of onset, patterns of clinical signs, rate of disease progression, and neuropathological changes. Mutations in at least 14 different genes underlie different forms of human NCL, 17 and mutations in at least 8 different genes underlie different forms of canine NCL. 1 –3,7,14,15,26,28 The differences in the causative mutations account for much of the heterogeneity in disease phenotype. Even different mutations within the same gene can result in a notable divergence in disease progression among affected people. 26 Based on these observations, the differences in the clinical and pathological presentations in these 3 cases of feline NCL could be accounted for by either differences in the gene affected or different mutations within the same gene. Considering the histories of the 3 cats examined in this study, it is unlikely that they were closely related. Thus, it seems likely that each case resulted from a different mutation. Differences in the ultrastructural appearance of the storage material between these cases are consistent with this conclusion.
The precise underlying etiopathogeneses of NCLs are currently unknown, but it has been accepted for some time that human NCLs are associated with defects in genes that code for proteins involved in lysosomal function. 11,17 Thus, the NCLs are typically classified as lysosomal storage disorders. To date, no gene defect has been recognized in feline NCL, and prior to the current report, only the CLN3 gene has been examined for possible mutations. 9 The single study examining the CLN3 gene did not uncover any mutations, consistent with the CLN3 gene analysis of the present study. In the cat that was evaluated in the present study, CLN3 did contain 1 sequence variant that predicted an amino acid change relative to the feline reference sequence. However, this variant was unlikely to be the cause of disease in this animal given that the cat was heterozygous for the variant. With a few rare exceptions, the NCLs in other species, including those resulting from CLN3 mutations, have been shown to be inherited as autosomal recessive traits, so it is likely that this is the case for the NCLs in cats as well. However, because no information was available about relatives of the cats evaluated in this study, we do not know with certainty that the diseases in these animals were recessive in inheritance. However, the predicted amino acid change in CLN3 occurs at a location in which the normal amino acid varies between species, making it unlikely that the amino acid at this location is critical to normal CLN3 protein structure or function. Although some mutations in some candidate loci have been ruled out as the cause of NCL in the 1 cat evaluated in this study, it is likely that there are multiple genetic bases for feline NCL, just as there are for humans, dogs, and sheep. Therefore, mutations in the candidate genes that were evaluated should still be considered in other cases of suspected feline NCL, particularly if the pattern of clinical signs and the histopathological lesions differ from that of cat No. 3 in this study. It is also possible that at least some forms of feline NCL result from mutations in genes that have not yet been associated with NCL. It was not until a mutation in ATP13A2 was found to be associated with NCL in Tibetan Terriers 7 that a neurological disease in people resulting from mutations in the same gene was also confirmed as a form of NCL. 5 Successful identification of causative mutations and clarification of feline-human NCL pathophysiological correlations will be required before cats can be considered viable models of human NCL.
This report emphasizes the need for thorough investigation of other suspected NCL cases to provide insight into and further facilitate the characterization and categorization of this disease in cats. An integrated, multilevel approach involving thorough clinical, histopathological, ultrastructural, and molecular investigation is essential in identifying the different forms and causative gene defects of feline NCL in this overtly heterogeneous condition. The discovery of causative mutations of feline NCL will be facilitated if cases can be identified among cats for which both pedigree and health information is maintained. The discovery of canine NCL mutations has been aided by the availability of pedigree and clinical history information as well as DNA samples from dogs of the same breed. As with dogs, an NCL mutation in a purebred cat is likely to be the same among all affected cats from the same breed. Therefore, if a candidate mutation is found in a single affected purebred cat, the relationship between the candidate mutation and the disease phenotype can be evaluated by association analyses between genotype and phenotype among cats of the same breed.
Footnotes
Acknowledgements
We thank Juyuan Guo for assistance with the molecular genetic analyses, Ronda Aho and Jan Shivers for their histological and immunohistochemical assistance, and Dean Muldoon and Don Ariyakumer for their electron microscopy assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported in part by the Batten Disease Support and Research Association. Dr Chalkley’s residency was partially supported by Pfizer, Inc. through the American College of Veterinary Pathologists/Society of Toxocologic Pathologists (ACVP/STP) coalition.