
Abstract
MicroRNAs (miRNAs) are a class of small, noncoding RNA that post-transcriptionally regulate protein expression. miRNAs are emerging as clinical biomarkers of many diseases, including tumors. The aim of this study was to investigate whether miRNA expression could vary in melanoma samples derived from formalin-fixed, paraffin-embedded (FFPE) tissues. The study included 4 groups: (1) 9 samples of oral canine malignant melanoma, (2) 10 samples of cutaneous malignant melanoma, (3) 5 samples of healthy oral mucosa, and (4) 7 samples of healthy skin. The expression levels of 6 miRNAs—miR-145, miR-146a, miR-425-5p, miR-223, miR-365, and miR-134—were detected and assessed by quantitative reverse transcription polymerase chain reaction (RT-qPCR) using TaqMan probes. Cutaneous canine malignant melanoma showed a decrease of the expression level of miR-145 and miR-365 and an increase of miR-146a and miR-425-5p compared to control samples. MiR-145 was also downregulated in oral canine malignant melanoma. The miRNAs with decreased expression may regulate genes involved in RAS, Rap1, and transforming growth factor β (TGF-β) signaling pathways, as well as upregulated genes associated with phosphatidylinositol signaling system, adherens junction, and RAS signaling pathways. In conclusion, miR-145, miR-365, miR-146a, and miR-425-5p were differentially expressed in canine malignant melanoma and healthy FFPE samples, suggesting that they may play a role in canine malignant melanoma pathogenesis.
Melanocytic tumors are relatively common in dogs and can arise at different sites, although they mainly affect the oral mucosa and the dermis. 17,28 Malignant melanoma is the most common melanocytic neoplasm in the oral cavity of dogs, and the most common oral malignant tumor of dogs. 42 Oral canine malignant melanoma has often an aggressive biological behavior, characterized by rapid invasion of neighboring structures and a high propensity for regional and distant metastasis, and it is therefore associated with a poor long-term prognosis. 28,42 Oral canine malignant melanoma resembles human malignant mucosal melanomas of the head and neck, which represent a fatal malignancy. 28 Conventional treatment for oral canine malignant melanoma involves surgical resection and/or radiation of the primary tumor, often resulting in efficient local tumor control, 5,32 while treatment of metastatic disease has shown little promise. 6,34,45
Cutaneous malignant melanoma is the third most common malignant skin tumor in dogs, 49 representing 27% of all canine malignant melanomas. 15 Cutaneous malignant melanoma is thought to have a less aggressive behavior than those of humans, although metastases are reported in up to 30% to 75% of the cases, potentially resulting in a poor prognosis. 7,43 Surgical excision with wide margins represent the treatment of choice. 8
Previous studies have investigated the correlation between patient survival and the clinicopathological variables, and they suggested that clinical tumor staging, location, completeness of excision, adjunctive treatment, mitotic index, Ki-67 index, level of infiltration, and cell pigmentation have a prognostic impact; however, results have been sometimes conflicting. 9,18,37,40,41,43,48 The molecular profile of canine melanoma has been only recently investigated. Comparison of the transcriptome profiles of canine cutaneous melanocytoma and melanoma identified 60 differentially expressed genes involved in collagen metabolism and extracellular matrix remodeling. 10,20
MicroRNAs (miRNAs) are a group of small RNAs, with around 19 to 25 nucleotides, resulting from cleavage of larger noncoding RNAs. They act as post-transcriptional regulators of gene expression. 4 MiRNAs have been associated with several molecular pathways, including modulation of proliferation, apoptosis, differentiation, and cell cycle regulation; thus, dysregulation of their expression may contribute to a variety of diseases and disrupt pathways of fundamental importance in development of neoplasia. 11 During tumorigenesis, some miRNAs that negatively regulate oncoproteins (ie, tumor suppressor miRNAs) are downregulated, while those negatively regulating tumor suppressor genes (ie, oncogenic miRNAs, or oncomiRNAs) are upregulated. 27,46 The involvement of miRNAs in melanoma pathogenesis has been demonstrated in both humans 1,13,19,39 and dogs. 29 –31,44 The identification of miRNAs associated with oral and uveal canine malignant melanoma has been investigated by Noguchi et al 29 and Starkey et al, 44 respectively, using a microarray hybridization approach. To our knowledge, few data are available on the expression patterns of miRNA in oral and cutaneous canine malignant melanoma when compared to healthy controls. The aims of the present study were (1) to screen by quantitative reverse transcription polymerase chain (RT-qPCR) the expression of a panel of miRNAs, previously demonstrated to be related to melanoma (miR-425-5p, miR-134, miR-145, miR-146a, miR-223-3p, and miR-365), 2,22,26,30,35,36,39,51 in a cohort of oral and cutaneous malignant melanoma, and (2) to carry out functional enrichment analysis of target genes and functional interaction network analysis, to identify pathways affected by the differentially expressed miRNAs.
Materials and Methods
Study Population
Histopathology samples of cutaneous (non-digital) and oral mucosal malignant melanoma were computer searched, including records from January 2012 to December 2013. Cases were considered eligible only if blocks were available for review and tumors had positive staining for Melan A and PNL-2 antibodies with immunohistochemistry (IHC). Patient data were collected both from the pathology submission forms and via telephone calls to the referring veterinarians. The formalin-fixed, paraffin-embedded (FFPE) samples that satisfied these criteria (19; 9 oral, 10 cutaneous) were enrolled and subsequently divided into 2 groups: cutaneous malignant melanoma and oral malignant melanoma. Sample details are described in Supplemental Table S1.
Histology and Immunohistochemistry
Cases selected were reexamined by a board-certified pathologist (L.R.) using hematoxylin and eosin–stained slides under a brightfield microscope. Tumors were examined for quality of fixation and for areas of necrosis, inflammation, or hemorrhage. The mitotic index (MI) was calculated as the total number of mitotic figures in 10, tumor-representative, 400× (ocular FN: 22; objective 40×/0.65) high-power fields (HPFs). For IHC, sections were dewaxed and subjected to antigen retrieval in Dako PT buffer high/low pH (Agilent Technologies, Santa Clara, CA, United States) using a computer-controlled antigen retrieval workstation (PT Link; Agilent Technologies) for 20 minutes at 98°C. Sections were then immunolabeled in an automated immunostainer (Link 48; Agilent Technologies), using primary antibodies against Melan A (mouse anti-human Melan A, clone A103, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA) and PNL-2 (mouse anti-human melanoma marker PNL-2, A4502, 1:400; Agilent Technologies), as previously suggested. 38 This was followed by a 30-minute incubation at room temperature with the secondary antibody and polymer peroxidase-based detection system (Anti Mouse/Rabbit Envision Flex+; Agilent Technologies). The reaction was visualized with diaminobenzidine (Agilent Technologies). Consecutive sections were incubated with murine subclass-matched unrelated monoclonal antibody, which served as a negative control. The positive reaction was represented by a distinct brown cytoplasmic reaction. A canine melanoma known to express Melan A and PNL-2 was used as positive control. Only melanomas with more than 10% of positive cells with either Melan A or PNL-2 were included in the study (Suppl. Fig. S1). 41
Control Population
FFPE samples of normal skin and oral mucosa originating from postmortem cases were used as negative controls representative for oral mucosa. Cases with no oral pathology and with nontumor-related cause of death were included.
MiRNA Extraction and Real-Time Quantitative PCR
Upon observation with a brightfield microscope, a 2-mm diameter area representative of neoplastic growth, with no areas of necrosis, hemorrhage, or inflammation, was selected and labeled on the histological slide (Suppl. Fig. S2a). The same area was then identified in the wax block and labeled (Suppl. Fig. S2b). Using a disposable 2-mm diameter biopsy punch with plunger (Miltex, Rietheim-Weilheim, Germany), the area of interest was sampled and extracted from the block (Suppl. Fig. S2c); this tissue core specimen was subsequently placed in an Eppendorf tube (Suppl. Fig. S2d) and used to extract small RNA.
Small RNAs were extracted from the core tissue specimens using the miRNeasy Kit for FFPE blocks (catalog number 217504; Qiagen, Milan, Italy). The Caenorhabditis elegans miRNA cel-miR-39 (25 fmol final concentration) (catalog number 219610; Qiagen) was used as synthetic spike-in control due to the lack of sequence homology to canine miRNAs. The RNA extraction was then carried out according to the manufacturer’s instruction. The reverse transcription was performed using the TaqMan MicroRNA Reverse Transcription Kit (catalog number 4366596; Applied Biosystems, Monza, Italy) using miRNA-specific stem-loop RT primers, according to the manufacturer’s instructions. Reverse transcription reactions were performed in 15-μl volume reactions containing 1.5 μl 10× miRNA RT buffer, 1 μl MultiScribe reverse transcriptase (50 U/μl), 0.30 μl 100 mM dNTP mix, 0.19 μl RNase Inhibitor (20 U/μl), 6 μl custom RT primer pool, and 3.01 μl nuclease-free water. The custom RT primer pool was prepared combining 10 μl of each individual 5× RT primer to a final volume of 1000 μl; the final concentration of each primer in the RT primer pool was 0.05× each. Then, 3 μl RNA was added to each RT reaction. Every RT reaction mixture was incubated on ice for 5 minutes, 16°C for 30 minutes, 42°C for 30 minutes, and then 85°C for 5 minutes.
The quantitative PCR (qPCR) experiments were designed following the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines. Small RNA TaqMan assays were performed according to the manufacturer’s instruction. The selection of miRNAs was based on previous publications in which these miRNAs were correlated to melanoma in dogs or humans. The selected primer/probe assays (Life Technologies, Monza, Italy) included cel-miR-39-3p (assay ID000200), hsa-miR-425-5p (assay ID001516), mmu-miR-134 (assay ID001186), hsa-miR-145 (assay ID002278), hsa-miR-146a (assay ID000468), hsa-miR-365 (assay ID001020), and hsa-miR-223-3p (assay ID002295). 2,22,26,30,35,36,39,51 Quantitative reactions were performed in duplicate in scaled-down (12-μl) reaction volumes using 6 μl TaqMan 2× Universal Master Mix II (catalog number 4440044; Applied Biosystems), 0.6 μl miRNA-specific TaqMan Assay 20×, and 1 μl RT product per reaction on an Eco Real Time PCR detection System (Illumina, San Diego, CA, United States). The standard cycling program was 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. Data were normalized relative to the expression of cel-miR-39. MiRNA expression levels were presented in terms of fold change normalized to cel-miR-39 expression using the formula 2–▵▵Cq. 24 Predicted targets consisting of significant up- or downregulated miRNAs were computationally retrieved from the TargetScan (http://www.targetscan.org/vert_71/) and miRWalk (http://mirwalk.umm.uni-heidelberg.de/) databases. The predicted targets of either up- or downregulated miRNAs identified by both databases were examined using DAVID bioinformatic tool (https://david.ncifcrf.gov/), in order to perform functional annotation and biological pathway enrichment.
Statistical Analysis
Statistical analysis was performed using XLStat (AddinSoft, New York, NY). Statistical significance was accepted at P ≤ .05. Data were tested for normality and homogeneity of variance using the Kolmogorov-Smirnov test. As data were not normally distributed, nonparametric statistical tests were applied. Kruskal-Wallis test was used to assess differences in miRNA concentrations between malignant melanoma groups and control groups. P values were adjusted using Bonferroni correction. Linear regression was used to investigate any relationship between differentially expressed miRNAs and age. Spearman’s ρ test was performed to evaluate possible correlations among miRNA expression levels, mitoses, melanophages, and melanocyte rates. Principal component analysis was performed to evaluate single correlations among miRNAs.
Results
Study Population and Tumor Histology
The 9 oral and 10 cutaneous malignant melanomas were from dogs with a median age of 9 years (range, 6–13) and predominantly of mixed breed (n = 4), with Rottweiler (n = 2) and Cocker Spaniel (n = 2) overrepresented among other breeds. Median mitotic index was 2.5 (range, 0.8–11.4).
miRNA Expression
To characterize the differences between groups, principal components analysis (PCA) on control and malignant melanoma groups was performed (Fig. 1). Data points with a higher correlation had a smaller degree of separation within the chart; a probable correlation was predicted if the factors were within 45° of each other. Healthy samples were separated from malignant melanoma groups in both skin (Fig. 1a) and oral (Fig. 1b) samples and were well correlated to each another. Cutaneous malignant melanoma samples were positively correlated; in oral malignant melanoma samples, miR365a and miR-145 were negatively correlated, and no correlation was observed between miR-146a and miR-425.
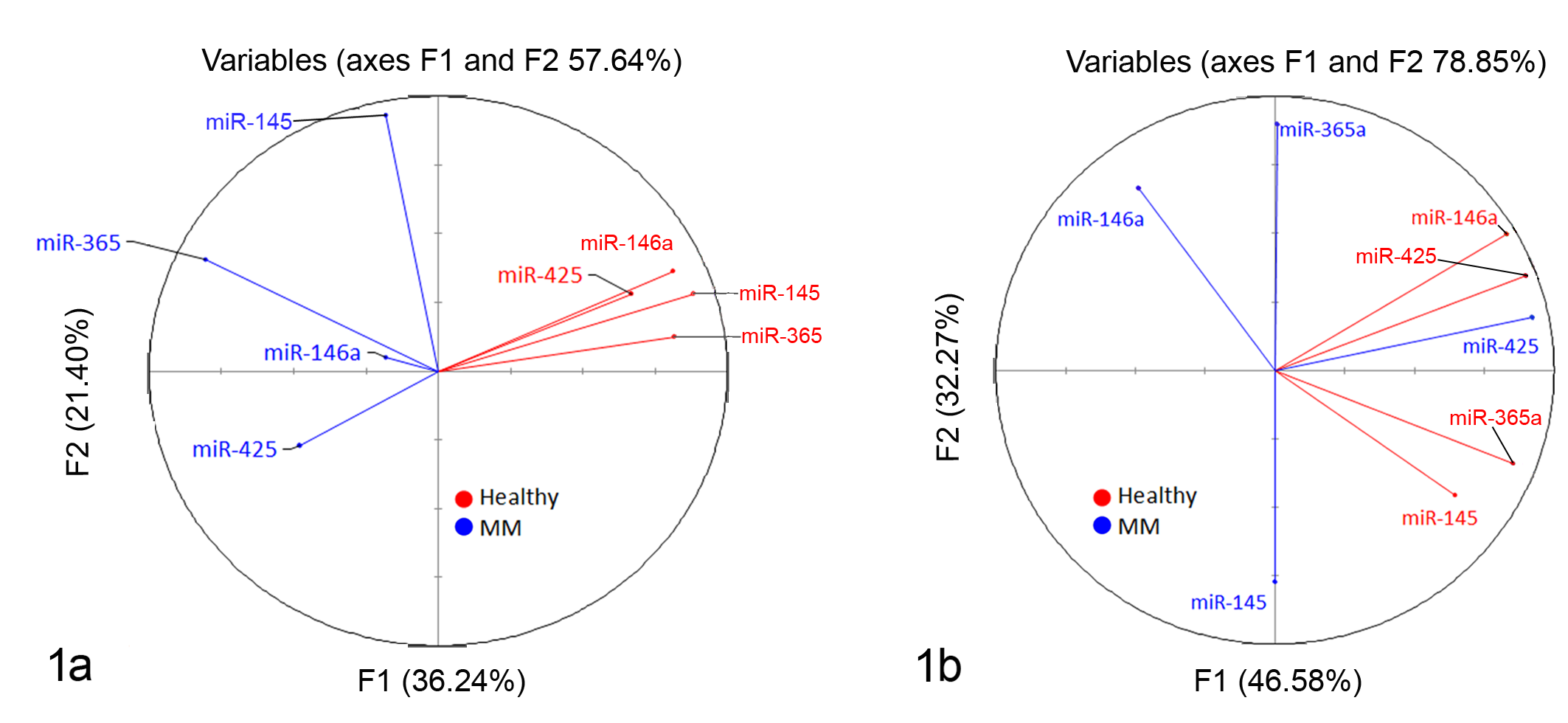
Principal components analysis of microRNA (miRNA) expression in canine malignant melanoma. The correlation circle shows the correlations between the miRNAs in (a) healthy skin and cutaneous canine malignant melanoma (MM) samples and (b) healthy oral mucosa and oral canine malignant melanoma samples. Variables with a higher correlation have a smaller degree of separation within the chart; a probable correlation can be predicted if the factors are within 45°. If 2 variables are far from the center and close to each other, they are significantly positively correlated; orthogonal variables are not correlated; and variables on the opposite side of the center line are negatively correlated.
The selected miRNAs were detected in all samples. Among these, miR-145-5p, miR-146a, miR-365, and miR-425-5p exhibited statistically significant differences among the malignant melanoma and control groups (Fig. 2). Compared to the corresponding nonneoplastic tissue, miR-145 (Fig. 2a) was downregulated in both cutaneous (P = .0076; ratio of cutaneous malignant melanoma/healthy skin = –5.7) and oral malignant melanoma (P < .0028; ratio of oral malignant melanoma/healthy oral mucosae = –7.6), miR-146a (Fig. 2b) and miR-425-5p (Fig. 2d) were upregulated, and miR-365 (Fig. 2c) was downregulated in cutaneous malignant melanoma (miR-146a: P = .02; ratio of malignant melanoma/healthy skin = 9.6; miR-425-5p: P = .04; ratio of malignant melanoma/healthy skin = 4.9; miR-365: P = .0087; ratio of malignant melanoma/healthy = –5.9), while there were no differences between oral malignant melanoma and control groups for these miRNAs. The expression levels of miR-223-3p and miR-134 were not different in malignant melanoma compared to healthy skin (Fig. 2e,f). The levels of differentially expressed miRNAs were not affected by the age of the dog (linear regression, P > .05). To test the possible collinearity, Spearman correlation analysis of miRNAs was performed, suggesting that there was a positive correlation among miR-145, miR-146a, miR-365, and miR-425-5p relative concentration (data not shown). No correlation was observed between miRNAs and mitotic count.
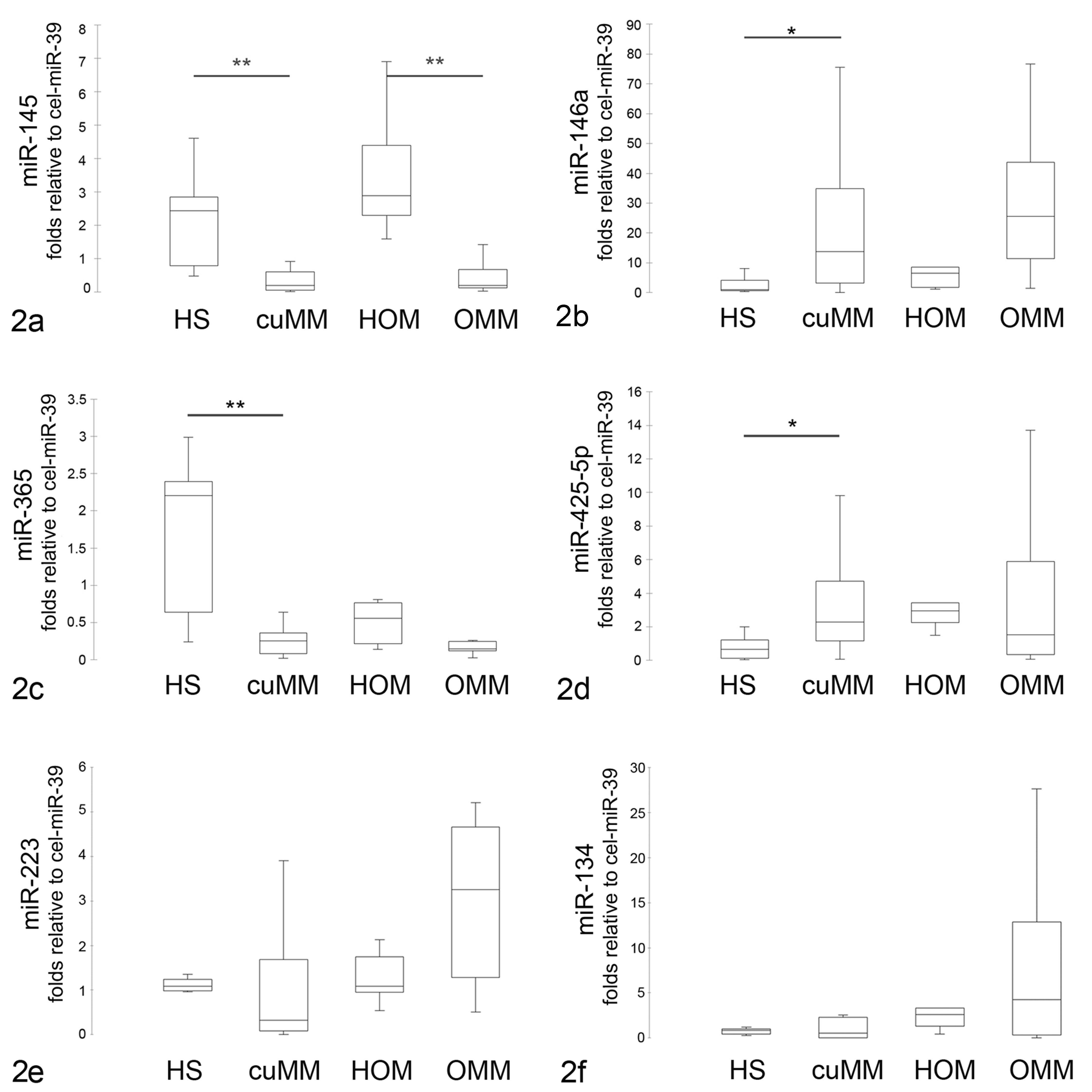
Expression of microRNAs (miRNAs) in canine malignant melanoma. Quantitative polymerase chain reaction results were normalized using cel-miR-39 as reference miRNA and the formula 2–▵▵Cq. Expression levels of miR-145 (a), miR-146a-5p (b), miR-365 (c), miR-425-5p (d), miR-223 (e), and miR-134 (f) in healthy skin (HS), cutaneous malignant melanoma (cuMM), healthy oral mucosa (HOM), and oral malignant melanoma (OMM). The boxes outline the quartiles, the horizontal line shows the median, and the whiskers show the range. *P < .05. **P < .01.
miRNA Target Prediction and Pathway Enrichment
To investigate the relevance to tumor development, predicted targets of either significantly up- or downregulated miRNAs were computationally identified by using TargetScan and miRWalk databases. The number of predicted targets shared by both databases included 602 for miR-145, 95 for miR-365, 103 for miR-146a, and 98 for miR-425-5p. The mRNA enrichment was performed using the DAVID bioinformatic tool. The Gene Ontology analysis was carried out using DAVID at 3 different levels: molecular function, cellular component, and biological process (Fig. 3). Most Gene Ontology molecular function items mainly included genes involved in the regulation of transcription and serine/threonine kinase activity for both up- and downregulated miRNAs. The enriched Gene Ontology terms in cellular component converged on genes associated with the cytosol, nucleus, and nucleoplasm for both up- and downregulated miRNAs. Downregulated miRNAs may modulate ruffle and focal adhesion, which are involved in cell migration, whereas upregulated miRNAs may regulate activin responsive complex, which is activated by transforming growth factor β (TGF-β) and acts primarily through Small mothers against decapentaplegic (SMAD) proteins. The biological process items focused on the modulation of protein binding and transcriptional activator activity for both sets of miRNAs. The downregulated miRNAs influence genes involved in the SMAD binding, RNA polymerase, and DNA binding activity; upregulated miRNAs genes are involved in the regulation of transcription. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was separately performed on the targets of the 2 sets of miRNAs. The most significantly enriched pathways were RAS, Rap1, and TGF-β signaling pathways for downregulated miRNAs and phosphatidylinositol signaling system, adherens junction, and RAS signaling pathway for upregulated miRNAs (Fig. 4).
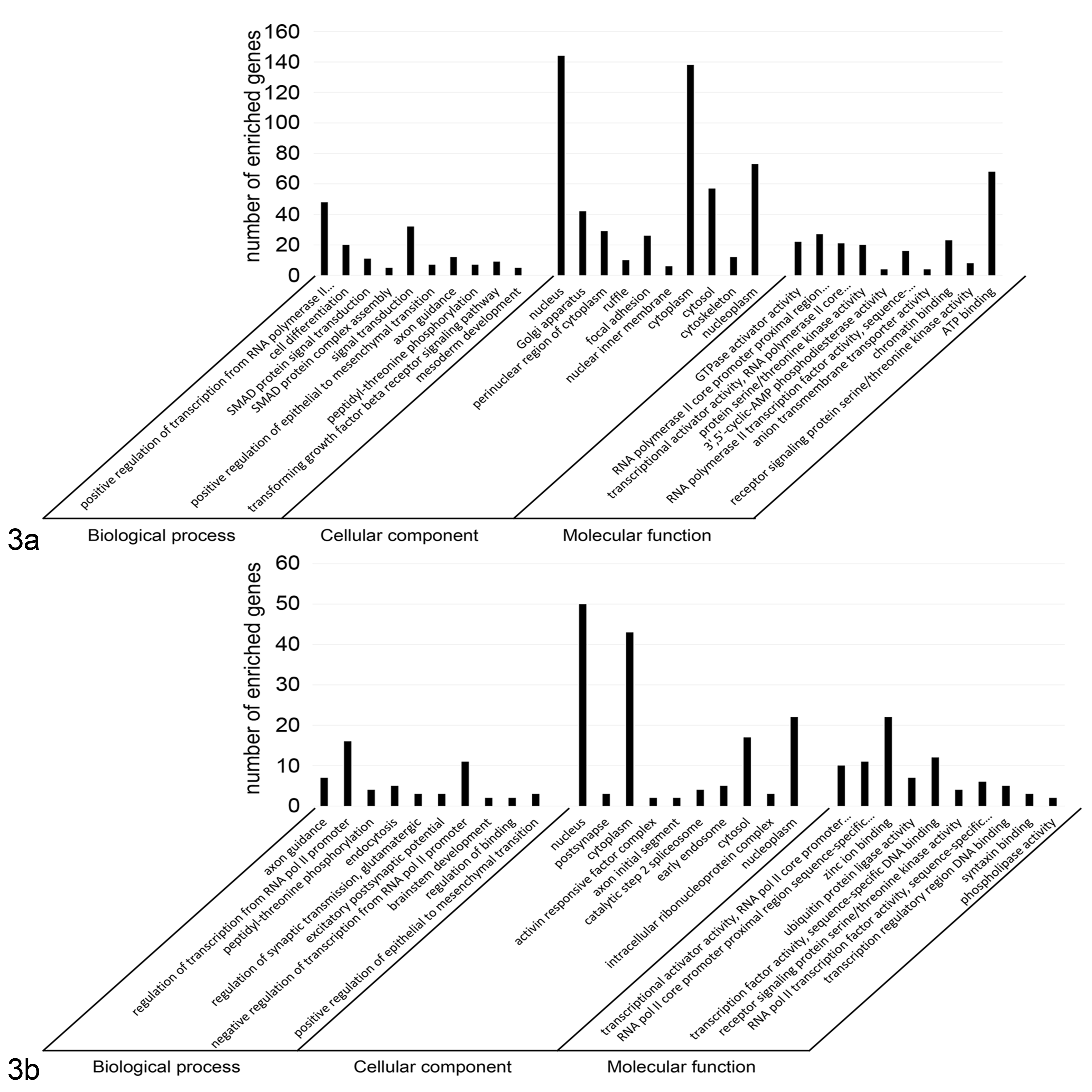
Gene Ontology annotation of genes predicted to be regulated by microRNAs (miRNAs) that were found to have significantly lower or higher expression in canine malignant melanoma samples compared to the corresponding healthy control tissues. The targeted genes were annotated by the DAVID tool at 3 levels, including biological process, cellular component, and molecular function. (a) Gene Ontology annotation of genes regulated by up-regulated miR-145 and miR-365. (b) Gene Ontology annotation of genes regulated by up-regulated miR-146a-5p and miR-425-5p.
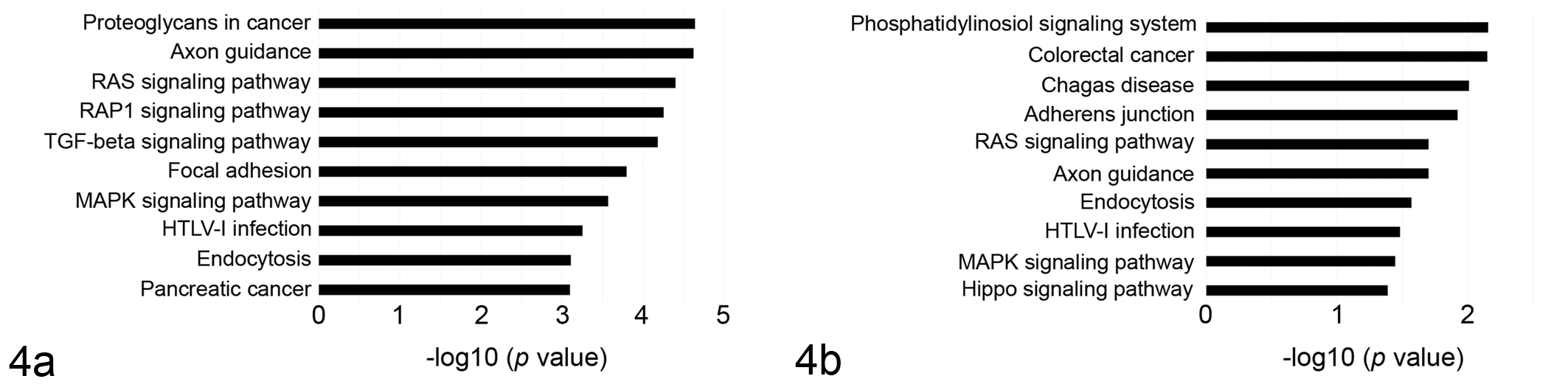
Pathway enrichment for microRNAs (miRNAs) that were found to have significantly lower or higher expression in canine malignant melanoma samples compared to the corresponding healthy control tissues. Genes were retrieved and enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway with DAVID tools. (a) Pathway enrichment for genes targeted by downregulated miR-145 and miR-365. (b) Pathway enrichment for genes targeted by up-regulated miR-146a-5p and miR-425-5p. The statistical significance level shown is the negative of the logarithm (base 10) of the P value.
Discussion
The role of miRNAs in canine malignant melanoma is only beginning to be defined. Using microarray and qPCR, Noguchi et al 30 analyzed the expression pattern of miRNAs in oral canine malignant melanoma and observed that miR-520c-3p was upregulated and that 6 other miRNAs (miR-126, miR-200a, miR-203, miR-205, miR-527b, and miR-713) were downregulated compared to healthy oral mucosa. Moreover, they demonstrated an association between the downregulation of miR-203 and shorter survival times. A recent study by Starkey et al 44 identified 9 miRNAs able to discriminate between metastatic or nonmetastatic canine uveal melanomas, therefore suggesting their potential in predicting biological behavior. The present study investigated the expression of 6 miRNAs in FFPE samples of oral and cutaneous canine malignant melanoma, and results suggest that specific miRNAs are differentially expressed in neoplastic vs normal tissue samples. Differentially expressed miRNAs identified herein have previously been implicated in human melanoma and other neoplastic conditions, as molecular regulators of tumor development and progression; a similar mechanism of action is hypothesized from the result of our study. MiR-145 was downregulated in both oral and cutaneous malignant melanoma, and so was miR-365 in cutaneous malignant melanoma. These were considered potential oncosuppressor miRNAs in accordance to the knowledge from the human oncology literature. It has been demonstrated that miR-145 and miR-365 modulate tumor cell growth, invasion, and metastasis by targeting NRAS, 23 c-MYC, 30,33 and neuropilin1, 2 respectively. The Gene Ontology and pathway analysis would suggest that miR-145 and miR-365 modulate cell migration and cell growth, influencing the RAS and RAP1 signaling pathways, among others. 3,23 Liu et al 23 demonstrated that expression level of miR-145 is lower in melanoma tissues than those in the matched adjacent normal tissues; conversely, NRAS levels are higher. A previous study also demonstrated that miR-365 influences the development of melanoma by targeting BCL2 and Cyclin D1, which are respectively involved in apoptosis and cell cycle progression. 53 We could speculate that the lack of regulatory miR-145 and miR-365 observed in oral and cutaneous malignant melanoma samples may be involved in a different aggressive behavior, which can be influenced by other molecular regulators.
MiR-425 and miR-146a were overexpressed in samples of cutaneous malignant melanoma. The Gene Ontology and pathway analysis showed that these miRNAs may target genes associated with cell proliferation, cell-cell adherens junction, TGF-β signaling pathway, and protein ubiquitination, which can contribute to tumor development and progression. In human medicine, miR-425 overexpression is associated with cell migration and invasion by targeting CYLD 50 and cell proliferation in gastric cancer, 52 hepatocellular carcinoma, 16 and esophageal squamous cell carcinoma. 22 The function of miR-425 in melanoma is still debated; Chen et al 12 demonstrated that the overexpression of miR-425/489 plays a pivotal role in the melanoma progression by activating the PI3K-Akt pathway. In contrast, Liu et al 21 suggested that miR-425 inhibits cell proliferation and metastasis, and it promotes apoptosis. The role of miR-146a has mainly been investigated in the context of immune response 47 while its role in human melanoma is still controversial. MiR-146a has been proposed as a negative regulator of immune response activation in melanoma by targeting STAT1 and IFN-γ in mice models, affecting melanoma migration, proliferation, and mitochondrial function as well as PD-L1 levels. 25 A recent study reported that miR-146a directly targets SMAD4, promoting cell metastasis and invasion; 35 Forloni et al 14 concluded that miR-146a plays a central role in the initiation and progression of melanoma by targeting NUMB, a suppressor of Notch signaling. Nonetheless, Raimo et al 36 hypothesized that miR-146a has 2 synchronous but distinctive functions in human melanoma: the enhancement of tumor growth and the suppression of cell metastasis.
Brachelente et al 10 characterized the transcriptome profiles of canine cutaneous melanocytoma and melanoma using a transcriptomic approach, identifying 60 differentially expressed genes. The comparison between this list and the list of genes potentially modulated by differentially expressed miRNAs identified in the present study showed that 4 genes (ADAM metallopeptidase with thrombospondin type 1 motif 2 [ADAMTS2], coiled-coil domain containing 80 [CCDC80], lysyl oxidase [LOX], and cysteine rich secretory protein LCCL domain containing 2 [CRISPLD2]) may be potentially modulated by miR-145 and miR-365 and one (SH3 domain GRB2-like endophilin interacting protein 1 [SGIP1]) by miR-146a and miR-425-5p. The RNA-seq results 10 showed that ADAMTS2, CCDC80, LOX, and CRISPLD2 are upregulated in cutaneous malignant melanoma, while SGIP1 is downexpressed; these results are consistent with the reduced or increased expression of molecular modulators, such as miR-145 and miR-365 and miR-146a and 425-5p, respectively.
In conclusion, the present study suggests that miR-145, miR-365, miR-146a, and miR-425 are abnormally expressed in cutaneous and oral malignant melanoma, potentially modulating pathways involved in cell proliferation. Further studies are needed to elucidate the real molecular targets of these miRNAs and to identify candidate targets for molecular therapies in the treatment of canine malignant melanoma.
Supplemental Material
Supplemental Material, DS1_VET_10.1177_0300985819868646 - MicroRNA Expression in Formalin-Fixed, Paraffin-Embedded Samples of Canine Cutaneous and Oral Melanoma by RT-qPCR
Supplemental Material, DS1_VET_10.1177_0300985819868646 for MicroRNA Expression in Formalin-Fixed, Paraffin-Embedded Samples of Canine Cutaneous and Oral Melanoma by RT-qPCR by Valentina Zamarian, Carlotta Catozzi, Lorenzo Ressel, Riccardo Finotello, Fabrizio Ceciliani, Miguel Vilafranca, Jaume Altimira and Cristina Lecchi in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.