
Abstract
Ductal plate malformations (DPMs) represent developmental biliary disorders with a wide phenotypic spectrum. This study characterizes DPM in 30 Boxer dogs. Median age was 1.5 (range, 0.3–10.0) years, with 12 dogs <1 year. Clinical features included increased serum levels of liver enzymes (28), gastrointestinal signs (16), poor body condition (14), abdominal effusion (9), and hepatic encephalopathy (2). Additional malformations included gallbladder atresia (8), atrophied left liver (2), absent quadrate lobe with left-displaced gallbladder (1), portal vasculature atresia (left liver, 1), intrahepatic portosystemic shunt (1), and complex intrahepatic arteriovenous malformation (1). All dogs had portal tracts dimensionally expanded by a moderate-to-severe multiple small bile duct phenotype embedded in abundant extracellular matrix; 80% displayed variable portal-to-portal bridging. Quantitative analysis confirmed significantly increased fibrillar collagen and a 3-fold increased portal tract area relative to 6 Boxer and 10 non-Boxer controls. Biliary phenotype was dominated by tightly formed CK19-positive ductules, typically 10 to 15 μm in diameter, with 3 to >30 profiles per portal tract, reduced luminal apertures, and negative Ki-67 immunoreactivity. CK19-positive biliary epithelium intersected directly with zone 1 hepatocytes as a signature feature when considered with other DPM characteristics. Phenotypic variation included a multiple small bile duct phenotype (all dogs), predominantly thin-walled sacculated ducts (4), well-formed saccular ducts (4), and sacculated segmental, interlobular, and intralobular ducts (Caroli malformation, 2 dogs, one with bridging portal fibrosis). Histologic evidence of portal venous hypoperfusion accompanied increased biliary profiles in every case. We propose that this spectrum of disorders be referred to as DPM with appropriate modifiers to characterize the unique phenotypes.
Keywords
Bile ducts and hepatocytes derive from a common precursor, the hepatoblast. Differentiation into biliary epithelium involves formation of the ductal plate, a composite formed by a double layer of embryonic epithelium surrounding a branch of the developing portal vein. 10,13 Remodeling by tubulogenesis defines and matures biliary epithelium into intrahepatic bile ducts. 10,13 This complex process is orchestrated by regulatory factors interacting in a time- and context-dependent manner. 57 Numerous transcription factors and genes are now recognized that influence epithelial, mesenchymal, and vascular proliferation; apoptosis; adhesion molecule expression; and cell polarity and orientation essential to this process. 51,57 Normally, ductal plate components not progressing through tubulogenesis undergo involution or become periportal hepatocytes. 6 Defective orchestration of epithelial-mesenchymal interactions during this process leads to retention of embryonic ductal structures or proliferative responses and excessive extracellular matrix that geometrically expand portal tracts. 57 Resulting abnormalities are collectively regarded as ductal plate malformations (DPMs). The most severe phenotype is associated with a multiple small bile duct phenotype with discontinuous biliary profiles embedded in abundant extracellular matrix that expands and bridges portal tracts (congenital hepatic fibrosis, CHF). The multiple bile duct phenotype represents changes during the developmental process and is not an active proliferative response as observed in ductular reactions. Small “hypoplastic” intrahepatic portal veins accompany most forms of DPM, resulting in diminished intrahepatic portal venous perfusion. 13 Whether portal veins are developmentally hypoplastic or merely reflect underperfusion and collapse as a result of the noncompliant exuberant extracellular matrix in the portal tract remains unresolved.
A wide phenotypic DPM spectrum is recognized in human beings and knockout mouse models. 57 Insufficient involution of embryonic ductal structures at the level of segmental bile ducts (first branch of the hepatic duct) can lead to formation of large dilated (cystic) ducts designated as Caroli disease. 13,14 Insufficient involution of ductal elements at the level of interlobular and intralobular bile ducts resulting in formation of large irregular saccular ductal elements and/or numerous small tightly formed ductal elements combined with abundant portal fibrosis is designated as Caroli syndrome (large duct involvement) or CHF (diffuse hepatic fibrosis leading to portal hypertension). 14,15 Milder phenotypes reflect failure to involute small ductal elements, resulting in retention of numerous tightly formed ductal elements with variable extracellular matrix that may or may not bridge between portal tracts. Isolated failure of ductal plate resorption at the level of interlobular bile ducts can lead to formation of Von Meyenburg complexes (isolated single microscopic lesions characterized by numerous tight bile duct profiles embedded in dense extracellular matrix). Diffuse formation of cystic biliary structures (mild to severe) is genetically more diverse and comparatively rare and can lead to polycystic liver disease. 57 Genetic mechanisms causing DPM implicate mutations affecting structure and function of primary cilia. 5,17,24,31,37,56,57 The abnormal bile ducts may reflect a differentiation defect, defective maturation of primitive ductal structures, or abnormal duct expansion or proliferation. 57 Primary cilia are solitary nonmotile organelles located on the apical surface of most mammalian cells. 5,57 These function as mechano-, osmo-, and chemoreceptors and are involved in designation of planar cell polarity, in cell cycle control, and in numerous additional signaling pathways. 57 Pathomechanisms leading to DPM remain elusive owing to the complexity of interactions that direct appropriate tubulogenesis. 17,24,31,37,57 Because primary ciliary dysfunction is a common factor linking DPM syndromes, they are now considered congenital or inherited cholangiociliopathies. 5,37
Human DPM syndromes are clinically and genetically heterogeneous, some manifesting shortly after birth and others recognized later in life. Late-onset syndromes are either serendipitously discovered or associated with acquired complications. Comorbidities reflect hepatic presinusoidal portal hypertension, development of acquired portosystemic shunts (APSS), predisposition for ascites, increased risk for bacterial infections of the biliary tree, and cholelith formation. Secondary conditions (infections, cholelithiasis, portosystemic shunting) may lead to episodic fever, lethargy, increased liver enzyme activity in serum, hyporexia, weight loss, abdominal distention, and rarely abdominal pain. 22,31,48 These features motivate clinicopathologic assessments, imaging studies, and liver biopsy.
Description of canine DPM associated with the CHF phenotype remained ill-defined until a recent study demystified its unique histologic characteristics. 5 Previously, dogs with this disorder had been confusingly intermixed with dogs thought to have portal vein hypoplasia or referred to as having hepatoportal fibrosis. 53,55 While a diverse spectrum of DPM has been described in various animal species, only a few appear to be familial, and until now, only a single breed-related rare kindred disorder has been described in more than 3 dogs, none without concurrent renal involvement. 18,19,23,25,38,40,54,58
After identifying DPM in 4 Boxer dogs presented to the Cornell University Hospital for Animals Teaching Hospital, a retrospective survey was undertaken to determine the frequency of this malformation in Boxer dogs in our anatomic pathology database (1990–2014). Upon identifying 30 dogs with this malformation, we designed a clinical and histomorphologic study to characterize DPM phenotypes within a single breed. We specifically sought to (1) characterize clinical, clinicopathologic, and survival features; (2) characterize histologic diversity; and (3) objectify the severity of portal tract expansion and hepatic fibrosis.
Materials and Methods
Case Selection
Using electronic databases and a series of search strings (biliary hyperplasia, bridging fibrosis, cirrhosis, bile duct obstruction, cholangitis and cholangiohepatitis, portal venous hypoperfusion) restricted to the Boxer breed, a search for accessions submitted to the section of Anatomic Pathology at Cornell University from 1990 to 2014 identified 138 cases. We identified 30 dogs (30/138) representing nearly 22% of Boxers with hepatic histopathology during this interval that fulfilled histologic criteria of DPM that included (1) expansion of portal areas with dense fibrous connective tissue; (2) increased small clustered biliary profiles, often with irregular contours (many lacking a visible ductal lumen, some were sacculated or oblong, with variable symmetry of duct epithelium [eg, irregular, thin, dysplastic appearance]) expanding portal tracts; (3) variable portal-to-portal bridging by ductal structures embedded in a dense fibrillar connective tissue matrix; (4) small-caliber bile ducts intersecting directly with hepatocytes through the limiting plate; (5) increased arteriolar profiles with thick muscular walls; (6) inconspicuous portal vein profiles; and (7) minimal portal inflammatory infiltrates. 2,57 Each feature was required for study inclusion with the exception of (a) portal-to-portal bridging, which was widely variable, and (b) 1 dog with a Caroli malformation that had intrahepatic cholelithiasis in sacculated ducts and a secondary Escherichia coli cholangitis. Of the 30 specimens that met inclusion criteria, 29 had been collected antemortem (20 surgical wedge biopsies collected during exploratory laparotomy, 8 laparoscopic biopsies collected using cup biopsy forceps, and 1 sample of 5 long cores collected using a 14-gauge tru-cut needle), and 1 sample had been obtained surgically at the time of death. The number of biopsies from different liver lobes and relative size of these biopsy specimens are provided in Supplemental Table S1. Six juvenile Boxers (3, 5, 21, 180, 270, and 360 days of age) and 10 adult non-Boxer dogs (4, 4, 5, 5, 8, 8, 9, 9, 10, and 14 years of age) were used as controls for histologic evaluations. The clinical histories of control dogs were reviewed to verify that reasons for tissue sample evaluation were unrelated to liver disease. Each control had been examined at necropsy or at surgery and had histologically normal liver based on examination of large liver samples. We sought a group of young Boxer dogs as controls because 12 of 30 (40%) cases were ≤1 year of age, and we reasoned that younger dogs might display portal tract features in transitional stages to adult morphology (eg, increased bile duct profiles). However, we also included a control group of older adult dogs to ascertain whether the young control Boxer dog population introduced bias. The portal tract area measurements and fibrillar collagen deposition for each of these groups were compared to Boxers with DPM and to each other.
Histology and Immunohistochemistry
Archived formalin-fixed, paraffin-embedded livers from cases and controls were sectioned at 4 μM and stained routinely with hematoxylin and eosin (HE), Masson’s trichrome, and rhodanine. HE-stained slides were used for morphometric studies of portal area dimensions. Masson’s trichrome-stained slides were used to assess the extent and distribution of connective tissue in portal tracts. Rhodanine-stained slides were used to assess the presence, zonal distribution, and severity of hepatocellular copper accumulation and, in some cases, to quantify hepatic copper concentration. For immunohistochemistry (IHC), 4-μM sections on positively charged slides were labeled with antibodies against cytokeratin 19 (CK19; Anti-Cytokeratin antigen [AE-1] clone ab74649, monoclonal; Abcam, Cambridge, Massachusetts), type IV collagen (Col-IV; Anti-Collagen IV antigen, clone ad6586, polyclonal, negligible cross reactivity with I, II, III, V, or VI collagen; Abcam, Cambridge, Massachusetts), and Ki-67 (Anti–Ki-67 antigen, clone MIB-1, monoclonal; Dako, Carpinteria, California) using a Dako autostainer plus and the streptavidin-biotin immunoperoxidase technique, according to manufacturer’s instructions. A duplicate of each section was incubated with a mouse isotype-matched immunoglobulin as a negative control. Positive canine tissues were concurrently stained with each set of slides. Three cases did not have rhodanine stains and 5 cases did not have IHC because paraffin blocks were unavailable. IHC for Ki-67 was done on tissue from 7 dogs, each with the typical DPM multiple small bile duct phenotype, and 1 with choleliths and E. coli cholangitis.
Digital images (400× magnification) of sections stained with HE, Masson’s trichrome, and rhodanine were captured using an Aperio ScanScope Digital Slide Scanner (Aperio Technologies, Vista, California). Using digital images of HE-stained liver sections, the circumference of the entire biopsy and all portal tracts were manually designated by an operator and areas of interest measured (μm2) using proprietary software algorithms (ImageScope software version 11.2.0.780; Aperio Technologies). Thereafter, the percentage of the biopsy area represented by portal tracts was calculated. Using digital images of Masson’s trichrome–stained liver sections, the relative amount of connective tissue was determined by color detection algorithms, using the same proprietary software. Image analysis was optimized for each slide by examining markup test images and individually adjusting hue width and saturation intensity limits to differentiate collagen deposition (blue color); the hue values ranged from 0.65 to 0.68. An algorithm was used to normalize the number of positive pixels ([positive + strong positive]/[(total positive + negative pixels) – (total number of weak positive pixels)]) to sample size. A similar procedure was used to quantify copper concentration, as previously reported, using an average hue value of 0.04. 9
Clinicopathologic Features
Medical records for each dog were reviewed and data extracted characterizing signalment, history, clinical signs, and clinicopathological parameters (complete blood cell count [CBC], serum biochemical profile, urinalysis, total serum bile acid concentrations); liver size judged from thoracic (disclosing liver margins) or abdominal radiographs, as well as abdominal ultrasound, computed tomography (CT) angiography, or gross inspection at surgery; and abdominal ultrasound features (hepatic echogenicity, parenchymal nodules, biliary structure dimensions, assessment of adequacy of intrahepatic and extrahepatic portal vein perfusion, and the presence or absence of gallbladder, abdominal effusion, and APSS). Suspected APSS were confirmed by color-flow Doppler ultrasonography, gross visualization during surgical or laparoscopic biopsy collection, isotope colorectal or splenoportal scintigraphy, or (in 1 case each) radiographic portography or CT angiography. Abdominal ultrasound examinations were completed by board-certified radiologists or experienced small animal internists. Referring veterinarians or owners of each dog were contacted by telephone to ascertain survival information.
Histologic Assessments
Histologic features were described based on inspection of slides stained with HE, Masson’s trichrome, and rhodanine. Features relevant to DPM were characterized by numerical scoring using tissues stained with HE and Masson’s trichrome and for immunoreactivity with CK19, Col-IV, and Ki-67 antibodies. Copper distribution was characterized for zone and number of hepatocytes demonstrating copper-protein cytosolic aggregates with severity designated by numerical scoring (0 = no copper, 1 = a few foci of hepatocytes [<2/10× field] inconsistently distributed in zone 3, 2 = consistent involvement of <25% of zone 3 hepatocytes [2–4/10× field], 3 = consistent involvement of >25% to 50% of zone 3 hepatocytes; 4 = consistent involvement of ≥50% to 75% of zone 3 and zone 2 hepatocytes, and 5 = panlobular involvement of most hepatocytes). 30
Statistical Analysis
Clinical data and image capture analyses data were examined for distribution using box-and-whisker plots and the Kolmogorov-Smirnov test. Because most data were nonparametric, details are presented as median and range. Dogs were stratified on the basis of APSS formation for comparison of clinical features between groups using 2-by-2 tables and Fisher’s exact test. Clinicopathologic data were defined respective to the reference range with the number of cases above or below the 95% confidence interval tabulated. Comparison of clinicopathologic parameters between dogs with and without APSS, as well as dogs stratified on the basis of copper determinations (cut-points of 400 μg/g dry weight liver [upper normal reference limit], 600 μg/g dry weight liver [used to determine clinical propriety of copper chelation in our hospital], and 1,000 μg/g dry weight liver [reflecting severe copper accumulation]), and comparison of the customized image analysis settings between cases and controls (hue value, hue width, hue saturation intensity) for collagen estimation were completed using the Wilcoxon rank sum test. Association between age and collagen accumulation based on image analyses was completed using Spearman rank correlation. Kaplan-Meier survival analysis (age) of dogs with and without APSS was done by using the Gehan-Wilcoxon test for short-term survival, the log-rank test for long-term survival, and the Cox-Mantel test and Peto-Wilcoxon test for overall survival. A value of P ≤ .05 (2-tailed) was considered significant for statistical comparisons, with the exception of clinicopathologic parameters where the P value significance was adjusted by Bonferroni correction to P ≤ .004.
Results
A primary diagnosis of DPM was recognized in 10 dogs, in 8 dogs with samples submitted for a second opinion, and in 12 dogs on reexamination of liver samples from dogs with initial diagnoses describing portal fibrosis, cholangitis or cholangiohepatitis, or portal venous hypoperfusion (Suppl. Table S1). The median (range) of age of dogs was 1.5 (0.3–10.0) years. Of dogs with DPM, the median age of those with and without APSS was not significantly (P = .98) different. No sex predisposition was recognized (15 males and 15 females).
Clinical Signs
Clinical history at presentation (Suppl. Table S2) included increased liver enzyme activity (28 of 30 cases), gastrointestinal signs (hyporexia [16]), vomiting [14], diarrhea [14]), difficulty gaining and maintaining body weight (14), abdominal effusion (9), and signs consistent with hepatic encephalopathy (2). Dogs with (n = 19) and without acquired portosystemic shunting (n = 11) did not display significantly different clinical features (all P > .25).
Clinicopathologic Features
Pertinent clinicopathologic features are provided in Table 1. A CBC was available from all but 3 dogs (dog Nos. 5, 6, and 26), and the serum biochemical profile was available from all but 2 dogs (dog Nos. 6 and 26). Four dogs were anemic (dog Nos. 4, 9, 15, and 19), 6 dogs had microcytic erythrocytes (dog Nos. 3, 15, 16, and 20), and 6 dogs (dog Nos. 1, 4, 11, 15, 25, and 27) had serum phosphorus above the reference range consistent with their young age. Subnormal total protein, albumin, and cholesterol concentrations were present in 3 (dog Nos. 4, 9, and 15), 4 (dog Nos. 4, 9, 15, and 17), and 3 (dog Nos. 4, 9, and 20), respectively. Many dogs had increased alanine aminotransferase (ALT, n = 11), aspartate aminotransferase (AST, n = 10), and alkaline phosphatase (ALP, n = 12). Three dogs lacked an increase in ALT activity (dog Nos. 8, 9), 5 dogs lacked an increase in AST activity (dog Nos. 7, 8, 10, 12, and 13), and 7 dogs lacked an increase in ALP (dog Nos. 3, 7, 12, 13, 19, 20, and 27). In most dogs, the fold increase in ALT activity was ≥3, reaching as high as 50-fold in 1 dog (with APSS = 2.5 [0.3–8.0], without APSS = 6.3 [1.1–50.1], P = .01). The fold increase in ALP activity was usually ≥2, reaching as high as 18-fold in 1 dog with cholangiohepatitis (with APSS = 4.0 [0.7–11.4], without APSS = 2.4 [0.5–18.2], P = .4). Seven dogs demonstrated increased γ-glutamyl transferase activity (dog Nos. 1, 2, 4, 10, 11, 18, and 25). Two dogs were hyperbilirubinemic (dog Nos. 1, 30), thought to reflect sepsis, one after enterotomy and intestinal resection to remove a foreign body (corn cob) and the other with suppurative cholangiohepatitis that also had an hepatic arteriovenous (AV) malformation. Total serum bile acids (TSBAs) were measured in 20 dogs; 11 dogs (1, 3, 4, 9, 15, 16, 17, 20, 21, 27, and 28) had increased concentrations (reference range <25 μM/L). Significant differences in clinicopathologic parameters between dogs with and without APSS only included the red blood cell (RBC) mean corpuscular volume (MCV) and TSBA concentrations, all P
Age and Relevant Hematologic and Serum Biochemical Parameters for Boxer Dogs With DPM, With and Without APSS.
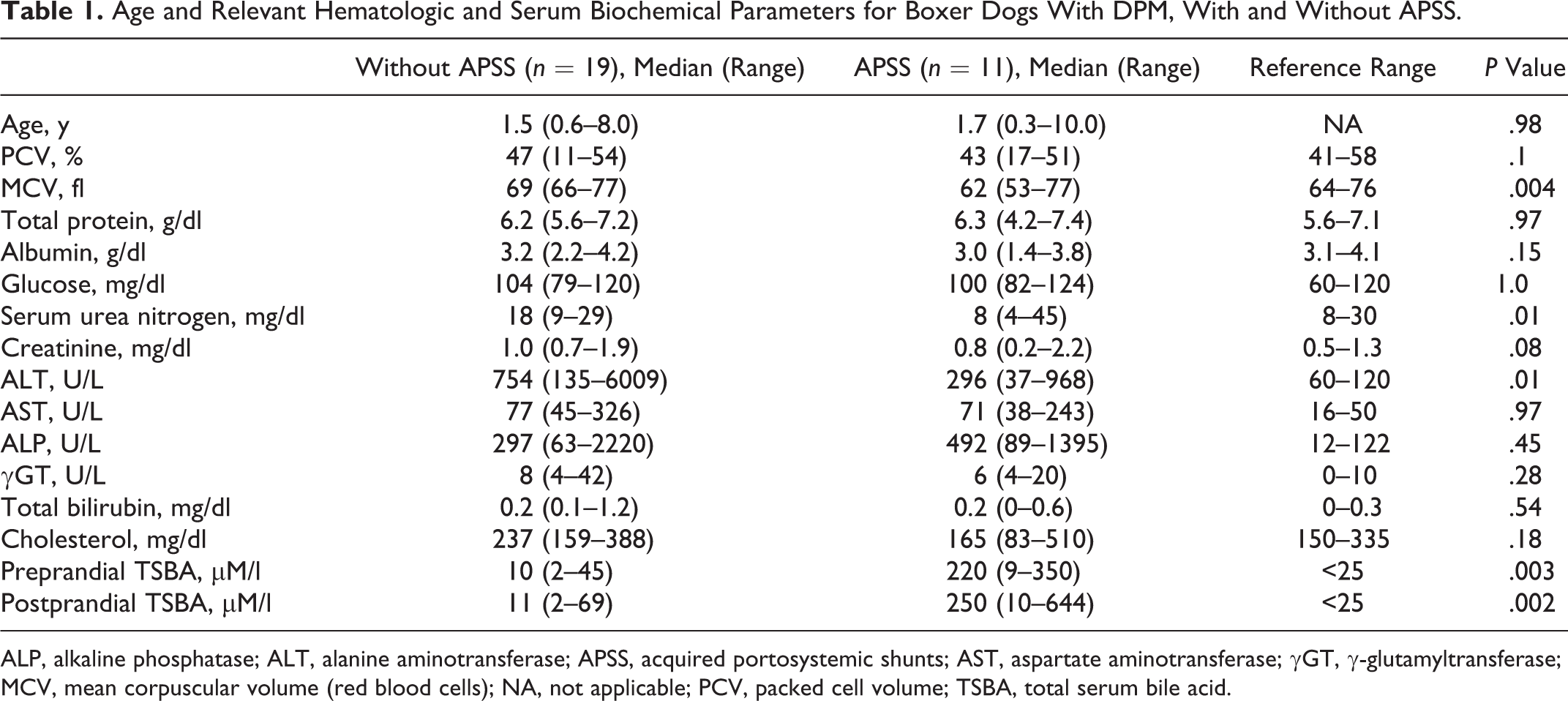
ALP, alkaline phosphatase; ALT, alanine aminotransferase; APSS, acquired portosystemic shunts; AST, aspartate aminotransferase; γGT, γ-glutamyltransferase; MCV, mean corpuscular volume (red blood cells); NA, not applicable; PCV, packed cell volume; TSBA, total serum bile acid.
Abdominal ultrasound examination, completed in 27 dogs, detected a small liver in 19, reduced intrahepatic portal flow (hypoperfusion) in 5, APSS in 8, a coarse or mottled liver echogenicity in 9, hypoechoic hepatic parenchyma in 2, inability to image a gallbladder in 8, abdominal effusion in 10, small urinary calculi in 4, and large duct intrahepatic choleliths in 1 dog.
Reconciling imaging and surgical assessments, 20 dogs were judged to have a small liver, 8 dogs judged to have a normal liver size, and 2 dogs judged to have a large liver. Each of 11 dogs with APSS had a small liver, with one having a congenital intrahepatic portosystemic vascular malformation and another with a congenital intrahepatic arteriovenous malformation. Eight dogs were confirmed to lack a gallbladder; 2 dogs were confirmed to have a markedly atrophied left liver, with 1 dog missing a quadrate liver lobe and gallbladder displaced within the left liver; and 1 dog had atretic portal vasculature within left liver lobes. No dog demonstrated individual large cysts within liver, pancreas, or kidneys on ultrasound imaging or during surgical biopsy, consistent with a polycystic disease syndrome. All 10 dogs developing abdominal effusion had APSS. Most effusions were characterized as modified transudates.
Sixteen dogs remained alive at the time of data analysis (without APSS n = 11, with 3 lost to follow-up censored from survival analysis; with APSS n = 5, no dogs lost to follow-up). The median survival was 8.8 years of age in dogs without APSS and 7.5 years of age in dogs with APSS. There were no significant differences in short-term, long-term, or overall survival between groups (Fig. 1, all P > .35), although there was a trend for shorter survival in dogs with APSS.
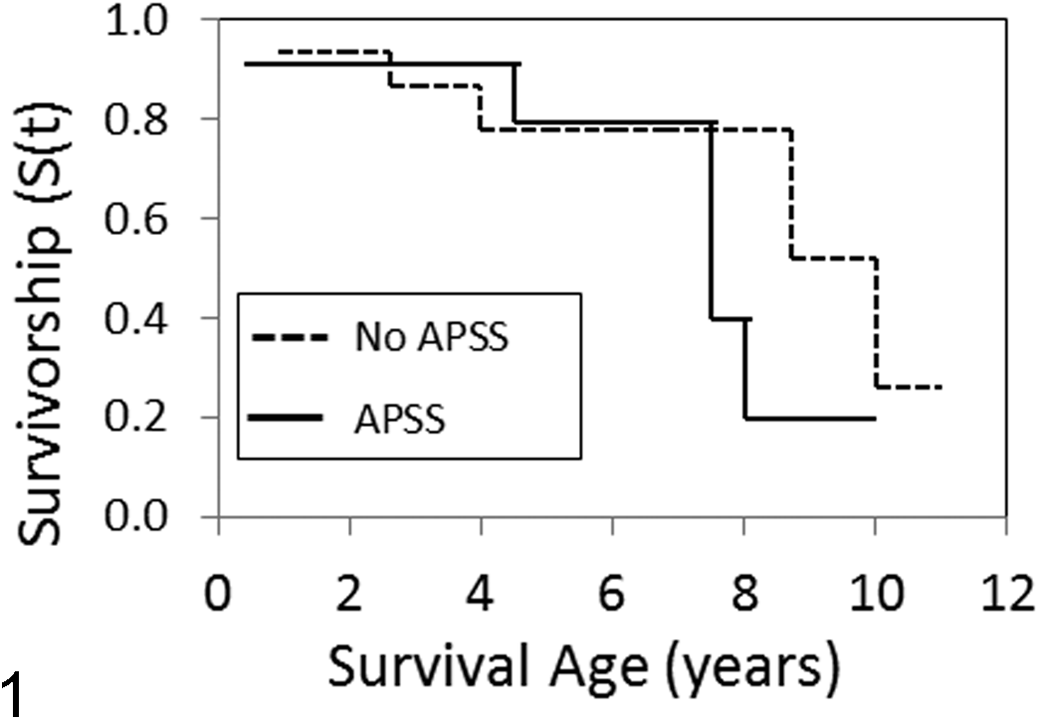
Kaplan-Meier survival curves for Boxer dogs with ductal plate malformations with (n = 11) and without (n = 19) acquired portosystemic shunts (APSS) showing survival as age at death (years). S(t) represents the survival probability function.
Histologic Evaluations
Twenty-four of 30 (80%) dogs had dimensionally expanded portal tracts with marked alteration in shape and moderate to severe bridging portal fibrosis (Fig. 2). Portal tracts were populated by numerous bile duct profiles with predominantly oval or circular shape ranging in diameter from 10 to 15 μm. The number of bile duct profiles widely varied from 3 to more than 30 per portal tract. Bile ducts in areas with the multiple small bile duct phenotype either lacked or had only a tiny luminal aperture. Most ductal epithelium was cuboidal, and most ducts were devoid of stainable intraluminal material or cellular debris and mitotic activity. In every case, there were numerous small-caliber bile ducts that occasionally intersected directly with zone 1 hepatocytes (Fig. 3); these cells were immunoreactive for CK19 antibody (Fig. 4). In addition to increased ductular profiles, portal tracts also were populated by oval to spindle cells that did not appear to form ductular or tubular structures. These cells were interpreted as either bipotential uncommitted progenitor cells (lacking CK19 immunoreactivity) or myofibrocytes (Fig. 4). While all dogs had well-formed ductular profiles with a multiple small bile duct phenotype, 4 also displayed sacculated bile ducts with irregularly angular lumens and thin duct epithelium, 4 displayed a combination of well-formed and saccular bile duct malformations, and 2 had saccular dilations involving segmental, interlobular, and intralobular bile ducts consistent with a Caroli malformation (Suppl. Table S3 and Figs. 5–7).
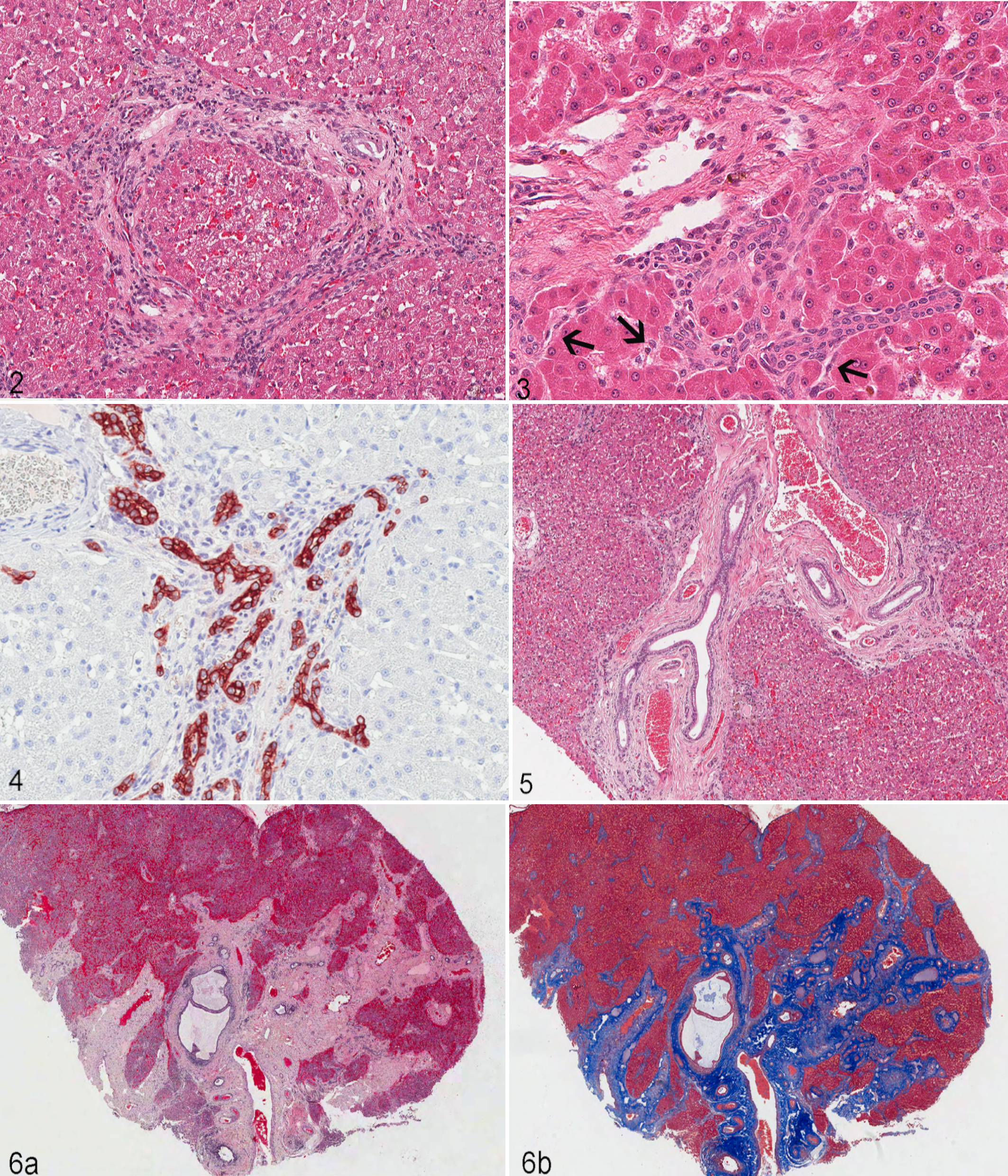
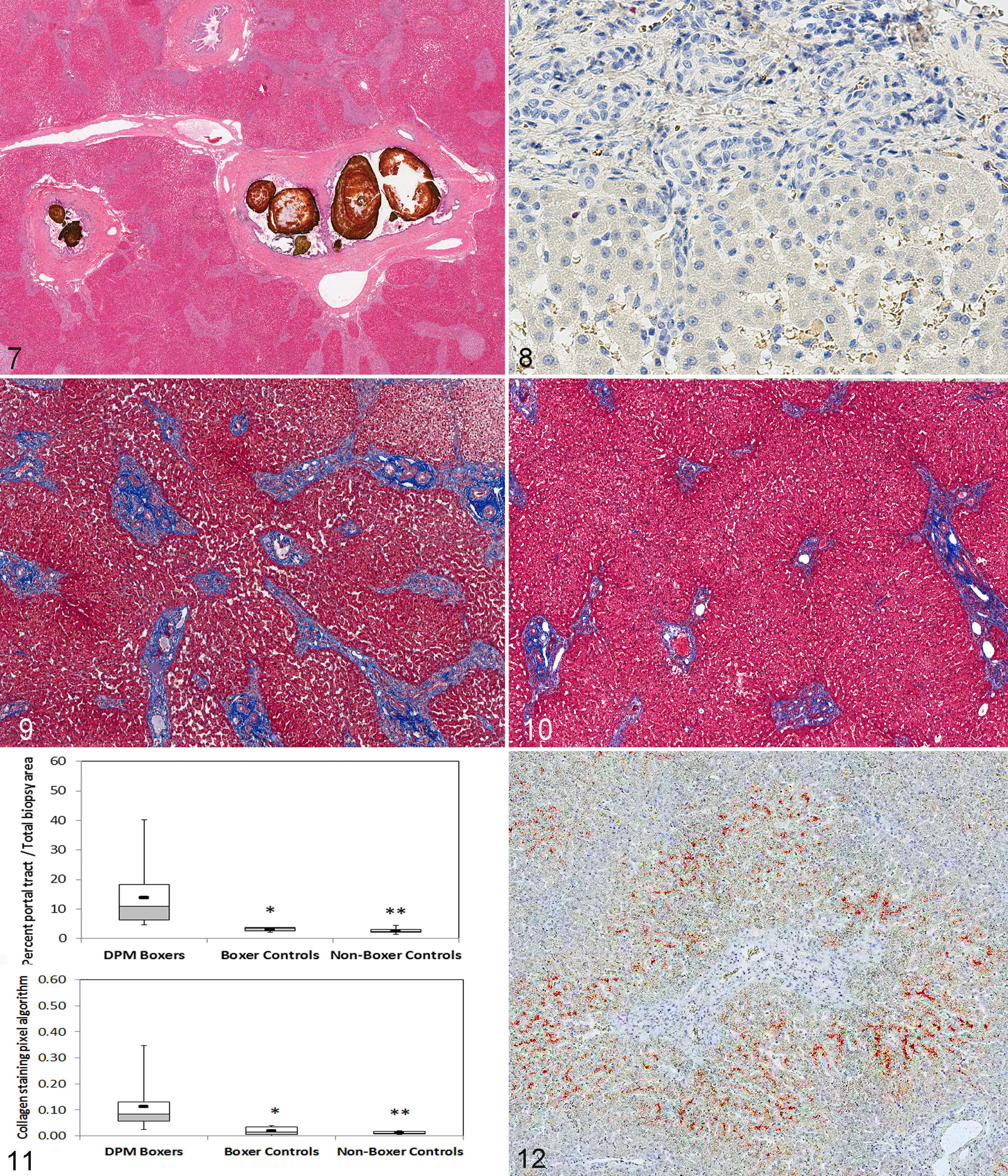
Active ductular proliferation was excluded by negative immunoreactivity in portal tracts for Ki-67 in 6 dogs (Fig. 8). Ki-67–positive immunoreactivity was present in portal tracts (Suppl. Fig. 1a,b) of 1 dog that had a Caroli syndrome complicated by an E. coli suppurative cholangitis and cholelithiasis. In this case, most of the Ki-67–positive cells had size and morphology consistent with infiltrating inflammatory cells. The Ki-67 staining also identified immunoreactivity in 0 to 7 hepatocyte nuclei per 400× magnification. Features consistent with portal venous hypoperfusion (diminished portal venous vasculature, hyperplastic arterioles) 45 accompanied biliary epithelial proliferation in every case. Portal tracts also often displayed small numbers of lymphocytes and plasma cells and, rarely, eosinophils, mast cells, and neutrophils. The number of inflammatory cells within portal tracts varied from 5 to 20 per 400× field. Cellular infiltrates in most cases were insufficient to determine if the inflammation was centered on portal connective tissue or on bile ducts. Lesions did not involve other acinar components (eg, central veins, hepatic cords) with the exception that islands of normal-appearing hepatic parenchyma were occasionally entrapped between bridging portal septa (Figs. 2, 3).
Image Analysis
Portal tracts in dogs with DPM had small, tightly formed bile ducts embedded in a moderate to severe conglomerate of dense extracellular matrix that was strongly positive for fibrillar collagen upon staining with Masson’s trichrome (Figs. 9, 10). There were no significant differences between customized image analysis settings between DPM and control dogs (hue [P = .44], hue width [P = .15], saturation [P = .69]). Summated median area of portal tracts, expressed as percentage of total biopsy area, was significantly higher in DPM Boxer dogs (11.0%, 4.6–40.2) compared to controls (Boxer controls: 3.5% [2.1–3.8]; P = .0004; non-Boxer controls 2.4 [1.5–4.4]; P = .00001). Notably, the median portal tract area was greater for all cases than for any of the controls (Fig. 11).
Positive pixel quantification of Masson’s trichrome–stained sections objectively confirmed significantly larger amounts of fibrillar collagen in DPM dogs compared to controls (Suppl. Figs. S2 and S3, Figure 11; P = .001 for Boxer controls, P = .00001 for non-Boxer controls). However, the relative amount of fibrillar collagen was widely variable among DPM dogs, with the values in 2 dogs overlapping the range found in controls. There was no association between severity of extracellular matrix deposition and age (P = .59).
Immunohistochemistry for Type IV Collagen
Mild to moderate positive immunoreactivity for Col-IV was observed diffusely in sinusoidal basement membranes in both cases and controls (Suppl. Fig. S4). Positive staining within the adventitia of portal tracts was evident in 50% of DPM dogs. Similar sinusoidal and portal tract staining patterns also were observed in control dogs and in the liver of an adult Boxer dog with cholangiohepatitis.
Rhodanine Staining for Copper
Rhodanine staining for copper was examined in 27 dogs and hepatic copper was quantified in 29 dogs (atomic absorption spectroscopy [n = 8], or digital analysis of rhodanine-stained sections [n = 21]). 9 Rhodanine staining confirmed cytosolic copper-protein aggregates in centrilobular hepatocytes in 23 of 27 (85%) cases (Fig. 12). Twelve dogs were estimated to have moderate to severe copper accumulation based on semiquantitative assessment scores (≥3 out of a 5-point scale). Median hepatic copper concentration of all dogs with measured copper (525 μg/g DWB [180–1594]) exceeded the upper limit of the reference range (>400 μg/g dry weight basis). Increased copper concentrations occurred in 17 of 29 (59%) dogs, with 10 of 29 (31%) having values >600 μg/g DWB (a cut-point for deciding on copper chelation when associated with increased ALT and evidence of centrilobular injury, S. A. Center personal communication) and 4 of 29 (14%) dogs having values >1000 μg/g DWB (considered severe). Mild centrilobular hepatitis was observed in 3 dogs and rare centrilobular single-cell necrosis of hepatocytes was observed in one dog with hepatic copper >1000 μg/g DWB.
Discussion
This clinical, clinicopathologic, and histologic study characterized a breadth of DPM phenotypes with varying degrees of hepatic portal fibrosis and types of bile duct malformations in dogs of a single breed. Before concluding that this study defines a unique breed-related DPM disorder, similar rigorous investigation of other dog breeds for DPM syndromes should be undertaken. In the population studied, we found no sex predilection and presume that dogs were not closely related as cases were identified over a 20-year span from a wide geographic region. Pedigrees from each dog, however, were not available. There was a single dog with a kindred history of “vascular” malformations but no confirmed histologic diagnoses of a DPM-like lesion in other related dogs. We approximate a 22% frequency of DPM among Boxers with liver histopathology in our database. Yet, the real prevalence of DPM in this breed remains unknown considering the breadth of phenotypes encountered, lack of clinical illness in some dogs, and problems with recognition of this recently described malformation. Indeed, 20 of the 30 cases initially had other pathologic diagnoses of portal fibrosis, cholangitis or cholangiohepatitis, or portal venous hypoperfusion, and DPM was only recognized as second opinion or on later case review. In addition, DPM was serendipitously discovered in some dogs when liver tissue was examined for a more primary health concern such as development of bacterial cholangitis or cholelithiasis, or because of ALT activity associated with a copper-associated hepatopathy. In one case, DPM was diagnosed at necropsy in a young Boxer euthanized because of progressive meningoencephalitis. The relatively high frequency of DPM among Boxers in our database suggests that this condition should be considered in Boxer dogs with increased liver enzyme activity, suspected hepatobiliary disease, portosystemic shunting, or abdominal effusion.
The spectrum of clinical, clinicopathologic, and histologic features described herein is consistent with DPM phenotype diversity described in humans. A small number of clinicopathologic features distinguished dogs with severe DPM phenotypes associated with portal hypertension and APSS formation. Dogs with APSS had significantly lower RBC MCV and significantly higher total serum bile acid concentrations than dogs lacking APSS. These features are commonly associated with portosystemic shunting in dogs. In addition, all dogs with APSS had some degree of peritoneal effusion, although only scant amounts were detected by ultrasound (US) imaging in 3 dogs. Clinical presentation primarily for signs of hepatic encephalopathy or abdominal distention due to ascites was rare, occurring in only 2 dogs with APSS.
Clinicopathologic features cannot uniquely discriminate dogs with DPM from dogs with other hepatopathies as the most common feature was increased ALT and ALP activities. Considering the young age of many dogs, increases in ALP activity may have reflected bone-associated isoenzyme in addition to hepatic sources. However, specific ALP isoenzymes were not characterized. Finding concurrently increased GGT and ALP implicated enzyme origin from biliary structures in 5 dogs. 8 The source of increased enzyme activity (ALT, AST, ALP, GGT) in DPM may include a variety of speculated causes, including focal inflammation associated with bile duct dilation, biliary proliferation, or dynamic extracellular matrix expansion of portal tracts, as has been speculated in humans, or the accumulation of hepatocellular copper in some dogs. Hepatobiliary bacterial infection was confirmed by culture in only 2 dogs; both were mildly hyperbilirubinemic. In each case, sepsis was considered an underlying cause of hyperbilirubinemia, although one of these dogs also had large intrahepatic choleliths and cholangitis. 12 Absence of hyperbilirubinemia and/or hypercholesterolemia in most DPM dogs correlates with the absence of histologic features consistent with obstructive or obliterative ductal disease or diffuse cholangitis.
Abdominal US imaging importantly contributed to DPM characterization, identifying a subjectively small liver in many dogs, a coarse parenchymal echotexture reflecting altered portal tract anatomy and fibrosis, abdominal effusion in dogs with APSS, gallbladder atresia, congenital intrahepatic vascular malformations, and left-sided liver lobe atrophy or malformation. Multiorgan polycystic malformation was ruled out by the absence of large cystic structures separated from the biliary tree or large cystic structures within pancreas or kidneys.
Predominant histologic lesions of DPM in Boxers included variable degrees of portal fibrosis and marked increase in biliary ductal elements with only minor and inconsistent portal inflammation. As proposed in humans, low-grade inflammation in portal tracts may reflect chronic intermittent bacterial infection. 47 Similar to the histologic variation observed in human DPM syndromes, marked variation in the severity of portal tract fibrosis was documented in these dogs. Unlike DPM in humans, we did not recognize overlap syndromes involving formation of small von Meyenburg complexes or development of choledochal cysts. Only 2 dogs demonstrated extrahepatic and intrahepatic bile duct dilation and sacculation consistent with a Caroli malformation.
The genesis of hepatic fibrosis in DPM remains enigmatic and in human patients is slowly progressive with aging. 24 However, we did not find a significant increase in hepatic collagen deposition associated with patient age. Sequential measurements with advancing age in an individual patient may be necessary to distinguish this process. In parenchymal liver disease, activation of hepatic stellate cells is central to fibrogenesis, with stellate cells transforming to the myofibrocyte phenotype under the influence of transforming growth factor (TGF)–β1 and matrix metallopeptidase 9 (MMP-9) released from activated Kupffer cells. 4,16 Conversely, fibrosis in DPM is thought to derive from resident portal myofibroblasts activated by exposure to locally produced connective tissue growth factors such as TGF-β. 42 –44 Retention of fibrogenic cytokines within poorly vascularized fibrotic portal tracts may augment their local effects. 42 Epithelial-to-mesenchymal transition of biliary epithelial cells to portal myofibroblasts (stimulated by TGF-β) is also hypothesized. 26,35 Transformation-associated secretion of TGF-β (TGF-β2) and upregulation of TGF-β receptors (betaglycan) on portal myofibroblasts are proposed to initiate and perpetuate an autocrine-like process that continues portal tract collagenization. 26,42
Novel hepatobiliary malformations identified in this case series include congenital absence of a gallbladder, left-sided atrophy or hypoplasia of the liver and vasculature, and large congenital intrahepatic vascular malformations. Congenital absence of the gallbladder is a rare anatomic malformation in the dog but was identified in 27% of these dogs. Our findings are unprecedented in the veterinary literature, but similar findings have been observed in humans. In humans, gallbladder atresia often accompanies other developmental anomalies, including formation of hepatic cysts; abnormalities of the portal vein (anomalous branching), hepatic artery, or intrahepatic biliary structures; hypoplasia of left liver lobes; absence of the quadrate liver lobe; and situs inversus. 29 Gallbladder atresia also has rarely been reported as an inherited familial trait, although gene mutations have not yet been characterized. 41
Liver sections from every DPM case demonstrated diminished or absent profiles of portal veins and increased arteriolar profiles in portal tracts. Use of CK19 IHC clearly distinguished biliary epithelium from arteriolar vascular profiles. Histologic features in DPM dogs are identical to those observed in dogs with any cause of portal venous hypoperfusion and include finding close approximation of portal and centrilobular regions (reflecting hepatic atrophy), small or nonidentifiable intrahepatic portal veins, and increased numbers of cross and tangential sections of hepatic arterials with mural thickening (smooth muscle). While the terminology of portal hypoplasia has often been used to describe morphologic changes reflecting reduced portal perfusion in humans and dogs with DPM, we posit that definitive diagnosis of portal hypoplasia in the circumstance of intrahepatic presinusoidal noncompliant fibrosis in the portal tract is impossible. Identical portal venous and arteriolar features are caused by any syndrome restricting hepatopetal portal circulation (ie, portal venous blood flow toward the liver). 32,33 Because extrahepatic portal veins lack valves, presinusoidal resistance imposed by abundant noncompliant portal tract fibrosis leads to hepatofugal circulation (ie, portal blood flow directed away from the liver), formation of APSS, and initiation of the hepatic arterial buffer response that induces physiologic hepatic arterial adaptations. 32,34 This compensatory arterial response induces formation of arterial twigs, vascular coiling, and thickening of the mural smooth muscle, resulting in the commonly observed formation of prolific stout arterials that interdigitate with juvenile ductal elements in DPM portal tracts. 34,49,59 The same features typify dogs with congenital portal venous hypoperfusion (eg, dogs with congenital portosystemic shunts or with congenital malformations of tertiary portal branches as shown in microvascular dysplasia) and in dogs with surgically created shunts between the portal vein and vena cava. 3,7,33,39 In DPM, it also is probable that intrahepatic presinusoidal portal hypertension caused by development of noncompliant portal tract fibrosis initiates physiologic adaptive perfusion through the arterial peribiliary plexus, contributing to the histologic arteriolar changes. 11,50 There were no histologic features suggesting an alternative cause of sinusoidal hypertension (eg, regenerative nodules, sinusoidal fibrosis, parenchymal remodeling) in any case. Primary or congenital portal hypoperfusion (congenital portosystemic shunts, microvascular dysplasia) should not be confused with DPM because these conditions lack portal tract fibrosis and the marked multiple small bile duct phenotype observed in this syndrome. However, 1 Boxer dog in this study had DPM and a congenital intrahepatic portocaval shunt, and another had DPM and a congenital complex arteriovenous malformation. The multiple small bile duct phenotype observed in DPM also should not be mistaken for acquired cholangiopathies or extrahepatic bile duct obstruction. We confirmed absence of proliferative activity in a subset of dogs by confirming lack of immunoreactivity with Ki-67, a marker of nonhistone nuclear protein expressed in all phases of the cycle except Go. 36 Acquired cholangiopathies are associated with inflammation centered on bile ducts and may also have epithelial destruction and periportal hepatitis, and they often display a ductular reaction, features not observed in DPM. Obstruction of large bile ducts induces a distinct cholestatic pattern associated with comparatively fewer biliary duct profiles with evidence of proliferation, duct dilation, duct tortuosity, periductal edema, circumferential peribiliary fibrosis, inflammation centered on bile ducts, and concurrent hyperbilirubinemia (eg, jaundice). Obstructive cholangiopathies also induce less extensive portal fibrosis than observed in DPM, and the portal fibrosis usually does not bridge between portal tracts unless obstruction is complete and chronic (months duration).
During early embryogenesis, primitive bipotential cells within the liver express CK19. 2 As development progresses, CK19 expression wanes as cells commit to hepatocyte differentiation or persists and becomes more prominent in cells committed to biliary differentiation. Biliary profiles in all dogs were strongly positive for CK19 expression with staining patterns that confirmed the abnormal topographic shift and migration of bile ductules characteristic of DPM. This marker simplified recognition of direct juxtaposition of differentiated biliary epithelium with hepatocytes at or beyond the limiting plate. This feature was so common that we now consider it a signature indication of canine DPM when considered in context of other features. It is important that this feature is not used as a diagnostic feature in isolation as dogs with necroinflammatory liver disease (eg, hepatitis) develop ductular proliferation within newly formed fibrous septa, at the interface between septal/portal areas, or within parenchyma, in close contact or actually continuous with surrounding hepatocytes. 28 However, in such cases, this lesion is associated with an active inflammatory process, parenchymal remodeling, and cell proliferation in adjacent hepatocytes, hepatic stellate cells, myofibrocytes, and hepatic progenitor cells. 46 Ductular activity in DPM is quiet with the exception of occasional low-grade cholangitis, and ductular profiles are embedded in abundant extracellular matrix, with ductular profiles extending beyond the margins of portal tracts where they may directly interface with hepatocytes. Bile ductules had no Ki-67 immunoreactivity in the typical cases in this study, confirming the absence of a proliferative ductular reaction. Thus, the “signature feature” we describe—direct juxtaposition of differentiated biliary epithelium with hepatocytes at or beyond the limiting plate—is only useful when considered in conjunction with other morphologic characteristics that define DPM: portal tract fibrosis, a crowded population of ducts with the multiple small bile duct phenotype and peculiar duct morphology with bile ducts embedded in abundant extracellular matrix, but an absence of necroinflammatory processes that provoke centrilobular collapse, fibrosis, remodeling, and regenerative nodule formation.
The CHF phenotype of DPM in humans is often associated with fibrocystic renal malformations. In the cases described herein, renal tissue was available from only 2 dogs examined by necropsy; neither dog had histologic renal lesions. In addition, none of 27 dogs undergoing comprehensive abdominal ultrasound evaluation demonstrated altered renal size, contour, or cystic lesions. Only 1 dog developed mild azotemia associated with dehydration and sepsis due to a perforated gastrointestinal foreign body; azotemia corrected with rehydration and surgical recovery.
Finding substantial hepatocellular copper accumulation in nearly 35% of dogs was not unanticipated because of the implicated increase in dietary exposure since 1997 that we have previously recognized in Labrador Retrievers and other breeds (S.A.C. and S.P.M., unpublished data). 21,30 We confirmed exclusive centrilobular distribution of accumulated copper in these dogs, consistent with findings in other dog breeds. 30,52 Studies in dogs and our observations suggest that dogs do not accumulate copper secondary to chronic parenchymal or cholestatic disease, as manifested in humans and cats. 1,27,52 Presumably, copper-associated liver injury resulted in increased serum ALT activity that was the stimulus for liver biopsy in 3 dogs that demonstrated centrilobular inflammatory infiltrates and occasional necrotic hepatocytes.
In conclusion, this study characterizes clinical, clinicopathologic, survival, and histologic features of a spectrum of DPM phenotypes in presumably unrelated Boxer dogs. Findings should help define the phenotypic breadth of canine DPM in other dog breeds. We propose that these malformations be referred to in general as DPM, conserving the term CHF for dogs with bridging portal fibrosis, evidence of presinusoidal portal hypertension, and APSS, and conserving the term Caroli malformation for dogs with saccular dilations involving segmental, interlobular, and intralobular large bile ducts. Caroli malformation in dogs does not necessarily involve the kidney. Of the population studied, only 11 of 30 (37%) fulfilled criteria for CHF and 2 of 30 (7%) fulfilled criteria for Caroli malformation. Clinical details of Boxers with DPM confirm that definitive diagnosis requires liver biopsy; can be accompanied by gallbladder atresia, left-sided liver atrophy or hypoplasia, and congenital vascular malformations; and can be complicated by portal inflammation, cholangitis and bacterial infection, cholelith formation, and concurrent but unrelated hepatocellular copper accumulation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.