
Abstract
The receptor tyrosine kinase (RTK) KIT is a major focus of current research into canine mast cell tumors (MCTs). Little is known about the role of other RTKs, such as vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptors (PDGFRs). These RTKs are dysregulated in many human and animal cancers and are key regulators of tumor angiogenesis. The aims of this study were to assess the expression and activation (phosphorylation) status of KIT, VEGFR2, and PDGFR (α and β) in canine MCTs and to examine associations with various clinical outcomes. c-KIT mutational status and KIT cellular localization pattern were also evaluated for these tumors. Twenty-seven MCTs, consisting of 5 subcutaneous and 22 cutaneous tumors, from 25 dogs were evaluated. MCT biopsies, cultured mast cells, and skin from the surgical margin were analyzed through Western blotting. MCT biopsies were also used for KIT immunohistochemical labeling and polymerase chain reaction for c-KIT mutational analysis. MCT had heterogeneous expression profiles for all 3 RTKs, which varied in intensity and activation status. Statistical analyses showed phosphorylated KIT, VEGFR2, and KIT cellular localization to be predictive of decreased survival time, disease-free interval, and increased metastatic rate. Expression of VEGFR2 and KIT diffuse cytoplasmic labeling were also significantly associated with increased rate of local recurrence. The results of the study show that phosphorylated KIT, KIT, VEGFR2, and PDGFRβ, in addition to KIT localization, may be valuable prognostic determinants in MCTs and should be further studied to improve diagnostic and therapeutic modalities.
Canine mast cell tumors (MCTs) are common skin tumors. 40 Although the majority are benign, some behave aggressively. It is difficult to identify the small number that are malignant with histopathology alone. 22,40 Aggressive variants do not respond well to chemotherapy and thus have a poor prognosis. 19,40 Consequently, there is a need to identify markers of aggressive MCT disease that can be targeted by novel therapies.
Receptor tyrosine kinases (RTKs) are cellular proteins that are intensively studied, as they are frequently dysregulated in human and animal neoplasia. 15,25 In nonneoplastic cells, RTKs are activated upon ligand binding. This results in receptor dimerization, phosphorylation of tyrosine residues, and activation of intracellular downstream pathways. 5 This culminates in the transcription of numerous genes, including those responsible for growth, survival, and motility. 5,41 In some types of neoplasia, RTKs are constitutively activated resulting in uncontrolled growth and tumor progression. 15,25 These overactivated RTKs are important therapeutic targets, as there are specific pharmacologic inhibitors of them. 15,24,25
In canine MCTs, the RTK KIT is a major focus of recent research. 13,24,33,39 Some canine MCTs have mutations in the gene encoding KIT (c-KIT) 11,20,43 that mimic those in human mast cell neoplasia (systemic mastocytosis) 13,38 and human and canine gastrointestinal stromal cell tumor. 14,35 In both mast cell neoplasia and gastrointestinal stromal cell tumor, c-KIT mutations cause uncontrolled tumor growth due to ligand-independent constitutive KIT signaling. 13,14,24
Canine MCT research led to the development of toceranib (Palladia; Pfizer, New York, NY, USA), a multitarget RTK inhibitor for use in dogs with MCTs. 24,27,33 In phase 3 trials, toceranib was clinically effective in dogs with MCTs having c-KIT mutations (60%) but also in those that did not harbor these mutations (>30%). 27 A prior study that supports these findings showed that toceranib caused regression of several types of canine primary and metastatic tumors, apparently without dysregulated KIT. 26 In these studies, only internal tandem duplication (ITD) mutations in exon 11 were evaluated. While it possible that other forms of KIT dysregulation were inhibited by toceranib (eg, other c-KIT mutations, autocrine or paracrine signaling, or spontaneous homodimerization of overexpressed receptor), an alternative possibility is that clinical responses may have been due to toceranib’s inhibition of RTKs other than KIT, such as vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR). 27 These RTKs are implicated in the progression of some animal 1,6,9,21,36,42 and human 4,7,8,16,18,32 neoplasms and are also key contributors to tumor angiogenesis. 12,17,29 Drugs that inhibit these RTKs could disrupt not only neoplastic cells but stromal interactions, vascularity, and metastatic niches. 12
The main objectives of this study were to determine the expression profiles and activation (phosphorylation) status of KIT, VEGFR2, and PDGFR for canine cutaneous and subcutaneous MCTs and to determine if they predict clinical outcomes. RTK expression in MCTs, normal skin at the surgical margins (used as a negative control), and cells cultured from tumors was evaluated to explore the relative distribution of RTKs within neoplastic cells and stroma. As c-KIT mutations and aberrant cellular localization (within neoplastic mast cells) play a significant role in some canine MCTs, 39,43 the presence of c-KIT mutations and KIT localization patterns were also determined for each MCT and assessed as risk factors for outcomes.
Materials and Methods
Case Selection
Twenty-seven samples of MCTs and normal skin from the surgical margins of 25 dogs diagnosed from 2006 to 2011 were obtained from the small animal surgical oncology service at the Ontario Veterinary College, University of Guelph, Guelph, Ontario, Canada. Five MCTs were histologically diagnosed as subcutaneous, and the rest were cutaneous.
MCTs were included if they met the following criteria: first, all were histologically diagnosed as cutaneous or subcutaneous MCTs; second, sufficient protein and genomic DNA could be collected from the sample; third, paraffin blocks were available for immunohistochemistry; and fourth, adequate follow-up data were obtained from veterinary clinics in the form of a questionnaire or telephone interview. MCTs were excluded from statistical analyses if there was no accompanying marginal skin available (n = 3); however, these were included for descriptive purposes. Follow-up information included signalment, tumor location, dates of prior or additional MCT development, metastasis, death or last examination, cause of death, and status at last examination. Additional information included details on adjuvant surgery or treatment (chemotherapy, radiation, and tyrosine kinase receptor inhibitors such as toceranib and masitinib).
The date of surgical excision was defined as the date of diagnosis. Follow-up time was the date of last follow-up or examination. Local recurrence was defined as regrowth at the surgical site, and distant occurrence was defined as occurrence of a subsequent cutaneous or subcutaneous MCTs at a different anatomic location. Metastasis to either local lymph node or widespread dissemination was determined by physical examination and at least 1 of the following: cytology of fine-needle aspirates, histology, radiographs, or ultrasound. Postmortem examination was not performed in any case. Disease-free interval (DFI) was defined as the time from date of diagnosis to the date of local recurrence, distant MCT occurrence, metastasis, or death, whichever occurred first. Dogs lost to follow-up, those still alive, or those that had died from causes unrelated to MCTs at the time of statistical analyses were right censored and included in the survival analyses. Median survival time was defined as the time at which 50% of dogs were alive, and median DFI was defined as the time at which 50% had no detectable MCT-related disease.
Tissue Processing and Protein Extraction
Within 30 minutes of surgery, tumor and marginal skin tissue were aseptically collected, minced, pulverized, and lysed for 1 hour at 4°C in protein lysis buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with 1mM PMSF, aprotinin and Phosphatase Inhibitor Cocktail 2 (2 μg/mL each; Sigma-Aldrich, Oakville, Canada), and 1mM sodium pervanadate (Thermo Fisher Scientific, Ottawa, Canada). Following centrifugation, aliquots were stored at –80°C. For 4 cases, tissue was collected as above and immediately stored at –80°C until protein extraction was performed.
For fresh MCTs, a portion was retained for in vitro cell culture. Tissue was aseptically minced and incubated at 37°C, in RPMI 1640 media (Sigma-Aldrich) containing 100 U/mL of sterile-filtered collagenase IV (Sigma-Aldrich) in a rotary shaker for 90 minutes. After digestion, cells were recovered through filtration via a 70-μm filter (Millipore, Temecula, CA, USA) and washed twice with phosphate-buffered saline containing 100 U/mL of penicillin, 100 µg/mL of streptomycin, 50 μg/mL of gentamycin, and 50 μg/mL of amphotericin B.
Fibroblast Coculture Method
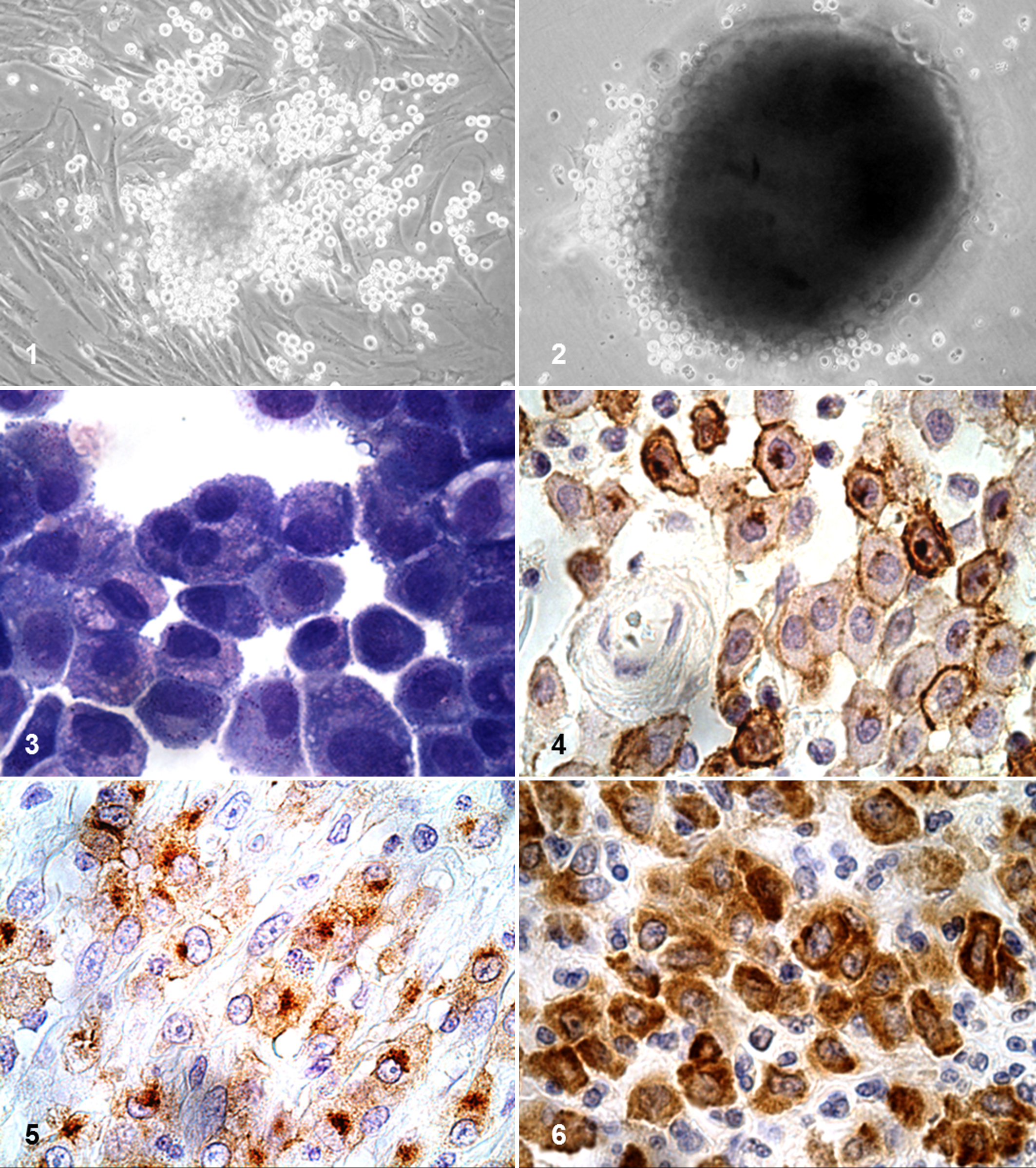
Cells were maintained in coculture with autologous fibroblasts through modifications of previously published techniques. 2,13,24 Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 via 6-well suspension plates (Thermo Fisher Scientific) in RPMI-1640 supplemented as described above plus 10% FBS, 1 mM sodium pyruvate, and 10 ng/mL of recombinant canine stem cell factor (R&D Systems, Minneapolis, MN, USA). Cell culture with suspension plates resulted in fibroblasts from tissue digest readily becoming adherent to plates; however, mast cells remained in suspension or attached to fibroblasts, forming spheroidal cellular aggregates (Figs. 1, 2). Media and recombinant canine stem cell factor were replaced every 3 to 5 days through gentle pipetting to avoid removing adherent mast cells. After 10 to 14 days in culture, a relatively pure (>90%) population of mast cells could be obtained by disaggregating mast cells from fibroblasts via more rigorous pipetting or 1% Vercene (Thermo Fisher Scientific). Purity was determined by cytology of Wright-Giemsa-stained cytospins (Fig. 3), and viability was determined with Trypan blue exclusion. To ensure that serum-derived growth factors were not present in the media, prior to all experiments, cells were washed and starved in serum-free medium for 48 hours with no additional recombinant canine stem cell factor. Cells were collected as described above, washed with phosphate-buffered saline, and lysed. For experiments involving phospho-VEGFR2, cells were incubated with 0.5mM sodium orthovanadate at 37°C for 8 minutes prior to lysis.
KIT Immunohistochemical Staining
KIT immunohistochemistry was performed with 5-µm-thick paraffinized tissue sections according to a previously published protocol. 39 Following deparaffinization, slides were incubated with 3% hydrogen peroxide to block endogenous peroxidases. Antigen retrieval was performed by heating slides in citrate buffer (pH 6, 1:10 of 10× Target Retrieval Solution; Dako, Carpinteria, CA, USA) for 30 minutes at 95°C in a water bath, followed by cooling at room temperature for 30 minutes. Slides were blocked with serum-free protein block (Dako) for 10 minutes prior to incubation with primary rabbit polyclonal antibody to KIT (1:100; Dako). A streptavidin–biotin–horseradish peroxidase labeling system (LSAB; Dako) was used for development, with 15-minute incubation times for the anti-rabbit biotinylated antibody and peroxidase-labeled streptavidin. Reactions were visualized with 3′,3′-diaminobenzidine (Dako). Following hematoxylin counterstaining, slides were mounted (Cytoseal; Thermo Fisher Scientific) and coverslipped. Negative controls were included for each tumor, and they consisted of canine cutaneous MCTs treated identically to the other tissue sections, except that antibody diluent (Dako) was used in place of primary antibody.
Evaluation of KIT Immunolabeling
KIT immunohistochemical labeling was evaluated in a blind fashion through published methods. 39 All slides were assigned 1 of 3 patterns of KIT protein localization as previously described by that study. KIT pattern I consisted of predominately membranous KIT protein localization with minimal cytoplasmic KIT protein localization. KIT pattern II consisted of focal to stippled cytoplasmic KIT protein localization and KIT pattern III consisted of diffuse cytoplasmic KIT protein localization. Cells on the margins of the tissue sections were not evaluated, owing to possible artifactual staining.
c-KIT Mutational Analysis
Mutational analysis was performed via previously published techniques. 11,20 Genomic DNA was extracted from frozen MCT tissue with a commercially available kit (DNeasy; Qiagen, Toronto, Canada), following digestion for 2 hours with proteinase K. Polymerase chain reaction amplification of c-KIT exon 11 and intron 11 was performed with a previously described primer pair 11,20 that flanks exon 11 and the 5′ end of intron 11, which includes the ITD region of the c-KIT proto-oncogene in canine MCTs; β-actin was used as a positive control (Table 1).
Primer Sequences for Polymerase Chain Reaction Detection of c-KIT Mutations.
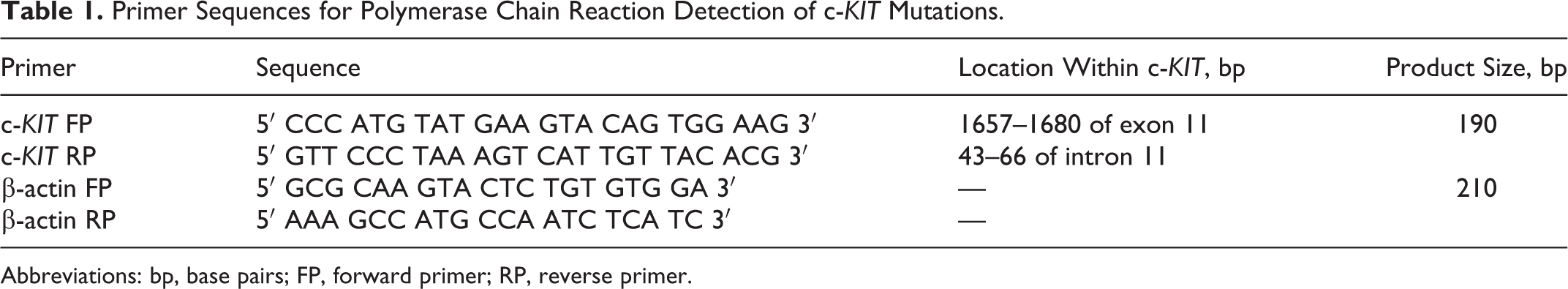
Abbreviations: bp, base pairs; FP, forward primer; RP, reverse primer.
Polymerase chain reactions were prepared in 25 µl of total reaction volume, with 10 pmol of extracted DNA, 7.5 pmol of each primer, 0.9 U of Taq polymerase (New England Biolabs, Whitby, Canada), 0.015 µmol of deoxynucleotide triphosphate, and 2.5 µl of reaction buffer (ThermoPol; New England Biolabs). Cycling conditions for c-KIT were as follows: 94°C for 4 minutes; 35 to 45 cycles at 94°C for 1 minute; 59.8°C for 1 minute; 72°C for 1 minute; and 72°C for 5 minutes. For β-actin, cycling conditions were 94°C for 4 minutes; 35 cycles at 94°C for 1 minute; 61.5°C for 1 minute; 72°C for 1 minute; and 72°C for 5 minutes. Amplified products were visualized by agarose gel electrophoresis on a 2% agarose gel after ethidium bromide staining. β-actin was used as a positive control, and negative controls were processed as above but did not contain template. Genomic DNA from samples with detectable c-KIT mutations was extracted through a commercially available kit (Ilustra GFX PCR DNA and gel purification kit; General Electric Healthcare, Mississauga, Canada), and sequencing was performed by the University of Guelph Laboratory Service.
Analysis of RTK Phosphorylation and Expression
Protein from tissues and cells was quantified (Bio-Rad protein assay; Bio-Rad, Mississauga, Canada), and 10 µg (for phospho-KIT and KIT detection) or 20 μg (for VEGFR2, PDGFRα and PDGFRβ detection) per sample of protein was loaded onto a 7% polyacrylamide gel. Sodium dodecyl sulphate–polyacrylamide gel electrophoresis was performed under reducing conditions, followed by protein transfer to a polyvinylidene difluoride membrane. Following blocking with 5% bovine serum albumin for 1 hour, membranes were incubated with anti-phospho-KIT (Tyr 719; Dako) or anti-phospho-VEGFR2 (Tyr 951; Cell Signaling Technology) at 1:1000 overnight at 4°C. For anti-PDGFRα and PDGFRβ (Tyr 849/847, 1:1000; Cell Signaling Technology) and anti-β-actin (1:2000; Cell Signaling Technology), blocking was performed with 5% milk prior to addition of antibody. After incubation with goat anti-rabbit POD-conjugated secondary antibodies (1:10 000; Sigma-Aldrich), membranes were successively rinsed and developed through BM Chemiluminescence Western Blotting Substrate (Roche Diagnostics, Mississauga, Canada) and exposed to x-ray medical film (Kodak Carestream; Sigma-Aldrich). Blots of phosphorylated proteins were then stripped (Reblot Plus Mild Stripping Solution; Millipore, Billerica, MA, USA) and reprobed with anti-KIT (1:2000; Dako), anti-VEGFR2, anti-PDGFRα, or anti-PDGFRβ (1:1000; Cell Signaling Technology). Positive qualitative controls were included for each experiment, consisting of canine cerebellum (phospho-KIT, KIT) and canine aorta (phosphorylated and total VEGFR2, PDGFRα, and PDGFRβ). A canine soft tissue sarcoma was used as a negative control for KIT and VEGFR2 and as an additional positive control for PDGFRα and PDGFRβ.
To confirm the identity of KIT, VEGFR2, and PDGFR and to ensure no antibody cross-reactivity, immunoprecipitation was additionally performed for these RTKs through MCT tissue and canine controls. For these experiments, approximately 1 g of protein lysate was incubated overnight with primary antibody at 4°C. The following day, 60 µl of sepharose G beads (Sigma-Aldrich) were added for 1 hour at 4°C. Following successive washes in 0.5% TNTE buffer, the supernatant was removed, and 60 µl of loading dye (Cell Signaling Technology) containing 2% β-mercaptoethanol (Sigma-Aldrich) was added to beads. Protein was eluted by incubating beads at 95°C for 5 minutes prior to electrophoresis, and Western blotting was performed as described above.
Densitometry of scanned blots was performed with Image J (National Institutes of Health, Bethesda, MD, USA) and analyzed with Prism 4 software (GraphPad, La Jolla, CA, USA). Relative phosphorylation was determined by normalizing phosphorylated protein levels with respect to total receptor values and β-actin (ie, p-RTK/RTK/β-actin), and relative total expression was normalized to β-actin (ie, RTK/β-actin), which was used as a loading control. Normalized densitometry values for paired MCTs and marginal skin samples were compared. MCTs having greater intensity of RTK expression compared with marginal skin were considered positive for expression, and those with lesser or equal expression were considered negative for expression. MCTs without available marginal skin were used for descriptive purposes only and were not included in statistical analyses.
Statistical Analyses
Risk factor analyses were performed with Cox proportional hazard models for the outcomes of survival, DFI, time to local and distant tumor occurrence, and metastasis. All risk factors, including demographic factors (ie, breed, sex, and age at diagnosis), were analyzed with univariable statistics. All statistical analyses were performed with SAS 9.1 (SAS Institute Inc, Cary, NC, USA). The correlation among risk factors was assessed via Spearman’s rank correlation coefficient based on the PROC FREQ option in SAS. A significance level of 5% was used for all statistical analyses.
Risk factors assessed were relative KIT phosphorylation, KIT, VEGFR2, and PDGFRβ expression, as well as histologic grade, mitotic index (MI), KIT pattern, and presence of an exon 11 c-KIT mutation. As there were only 5 cases of subcutaneous MCTs, separate analysis was not performed, so risk factor statistics are for both cutaneous and subcutaneous MCTs combined. For statistical analyses and descriptive purposes, grade was dichotomized into high (which included grade III cutaneous and high-grade subcutaneous MCTs) and low (cutaneous grades I and II and low-grade subcutaneous MCTs). Histologic grade was based on previously published grading schemes. 30,37 Results were also compared with the recently published 2-tier grading system that categorizes cutaneous MCTs into low- and high-grade tumors. 22 MI was defined by the number of mitotic figures per 10 high-power fields as previously published. 22 For analyses, MI was modeled as a continuous variable. KIT pattern was dichotomized into pattern III (diffuse cytoplasmic) as compared with combined patterns I and II, given that no statistical difference was seen between these patterns (data not shown). As there were so few dogs in each breed category, Labrador Retriever, Golden Retriever, mixed breed, Sharpei, and Boxer were separately compared with all other breeds for statistical analyses. Labrador Retriever compared with all other breeds is shown as an example.
The small sample size did not allow for multivariable model construction, as overfitting was inevitable. Also, as many of the variables were highly correlated (>60%), collinearity issues would have prevented many markers from being included in the same model. 10 Results are reported as hazard ratios: values for categorical risk factors are interpreted as the ratio of the predicted hazard of 1 group relative to a referent group (eg, presence vs absence of risk factor). The Cox proportional hazards assumption was graphically assessed through log-cumulative hazard plots and examination of Schoenfeld residuals. Evaluation of the functional form of the relationship between continuous predictors (age and MI) and clinical outcomes was assessed with Martingale residuals generated from the null model and plotted against the predictor, via a smoothing function (Lowess curve). Predicted survival curves were generated from univariable statistics through the baseline option in PROC TPHREG in SAS.
Results
Demographics
Tumor tissue and follow-up data were available for 27 MCTs from 25 dogs. Two dogs had local recurrences, and both original and recurring MCTs were used in the study, although only the original MCTs for each was included for statistical analyses. Marginal skin was not available for 3 cases, so these were used for descriptive purposes only; thus, 22 MCTs were included in statistical analyses. There were 15 breeds in the study, including Labrador Retrievers (n = 5), Golden Retrievers (n = 3), mixed breed dogs (n = 3), Sharpeis (n = 2), Boxers (n = 2), and 1 each of 10 other purebred dogs. The mean and median age was 7.1 and 7.6 years, respectively (range, 4 months to 12.3 years). There were 16 spayed and 9 neutered dogs. Breed, age, and sex were not found to be statistically significant risk factors for any outcomes (Table 2). Tumors occurred on the extremities (n = 10), trunk (n = 7), head/neck (n = 4), inguinal/perineal (n = 3), and abdomen (n = 1). Three dogs presented with 2 separate MCTs, and 2 dogs had concurrent osteosarcoma (censored). Only 1 MCT from dogs with multiple MCTs was included in the study, and all were diagnosed histologically with identical grades. Seven dogs (28%) had prior surgery for MCTs: 2 of these were local recurrences, and the remainder occurred at distant sites. In the surgery report, surgical margins were defined as “complete” according to 2- to 3-cm margins and a fascial plane deep. Surgical margins were complete in 76% of cases (19 of 25). Ten dogs received chemotherapy: 6 of these additionally received toceranib or masitinib, and 2 of these dogs were also treated with radiation. No dogs included for statistical analyses received adjuvant treatment prior to surgery. Of the 5 dogs with subcutaneous MCTs, 4 were Labrador Retrievers, and the last was a mixed breed. Subcutaneous MCTs occurred in the axilla, forelimb, flank, hip, and inguinal locations. Of these, 2 were female and 3 were male, ranging in age from 1.7 to 8 years.
Univariable Cox Proportional Hazard Analyses for Clinical Outcomes for 22 Dogs With Cutaneous and Subcutaneous Mast Cell Tumor.a
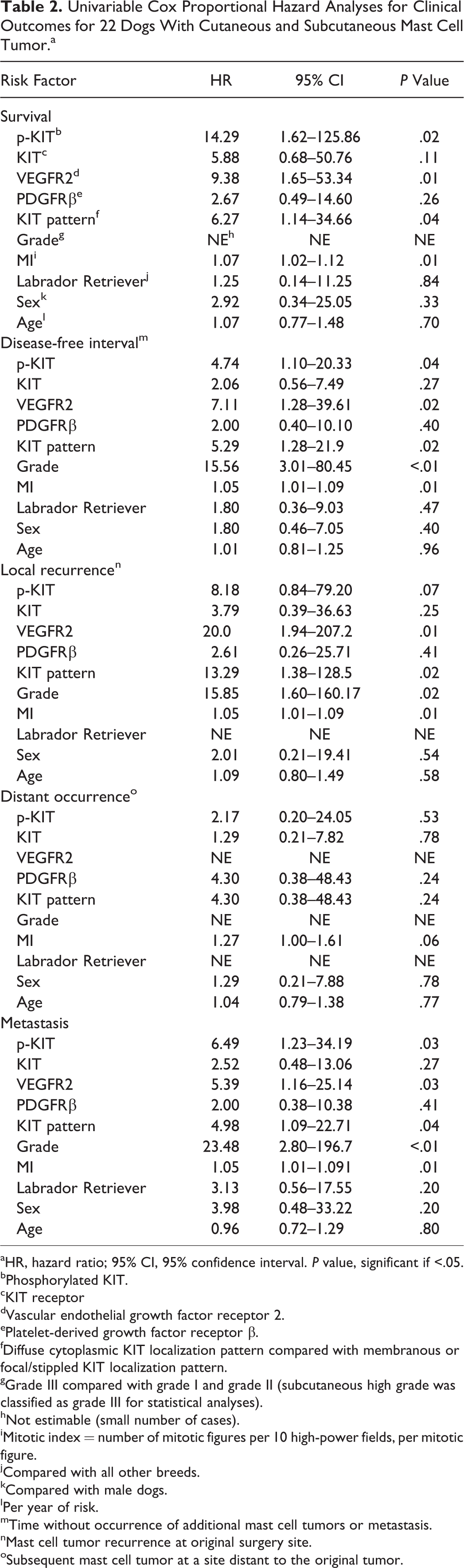
aHR, hazard ratio; 95% CI, 95% confidence interval. P value, significant if <.05.
bPhosphorylated KIT.
cKIT receptor
dVascular endothelial growth factor receptor 2.
ePlatelet-derived growth factor receptor β.
fDiffuse cytoplasmic KIT localization pattern compared with membranous or focal/stippled KIT localization pattern.
gGrade III compared with grade I and grade II (subcutaneous high grade was classified as grade III for statistical analyses).
hNot estimable (small number of cases).
iMitotic index = number of mitotic figures per 10 high-power fields, per mitotic figure.
jCompared with all other breeds.
kCompared with male dogs.
lPer year of risk.
mTime without occurrence of additional mast cell tumors or metastasis.
nMast cell tumor recurrence at original surgery site.
oSubsequent mast cell tumor at a site distant to the original tumor.
Clinical Outcomes
Over the study period (2006–2011), 12 dogs died, 9 to MCTs and 3 to unrelated causes. Deaths not due to MCTs were censored, as were dogs that were alive (n = 9) or lost to follow-up (n = 4). Of the 20 dogs with cutaneous MCTs, 10 dogs died, 8 to MCTs and 2 to unrelated causes. Of the 5 dogs with subcutaneous MCTs, 1 died from MCT disease (metastasis) 83 days after surgery, 1 to unrelated causes, and the other 3 were alive.
Local recurrence was reported in 6 dogs; 2 of these had tumors with incomplete surgical margins. Six dogs developed a subsequent MCT at a distant site, 1 of which had a low-grade subcutaneous MCT. The mean and median follow-up time for all dogs was 419 and 321 days, respectively (range, 10–1620 days), and for dogs with subcutaneous MCTs, this ranged from 83 to 999 days. Metastasis occurred in 9 (36%) cases, consisting of lymph node (n = 5) or disseminated spread (n = 4). Of these, 2 were subcutaneous MCTs: 1 dog developed local lymph node metastasis and died from MCTs, and the other, a 1.7-year-old Labrador with an inguinal MCT, had splenic metastasis at the time of initial surgery. The latter dog was treated with splenectomy, systemic chemotherapy, and toceranib and was alive at 515 days. Aside from that case, no other dogs had metastasis at the time of surgery.
Histologic Grading and MI
Cutaneous MCTs were histologically diagnosed as grade I (n = 7), grade II (n = 5), and grade III (n = 8), as assessed with a 3-tier system. 30 Based on a recently published 2-tiered system, these were diagnosed as low grade (n = 12) and high grade (n = 8). 22 There was no significant difference between grade I and II tumors, so they were combined for comparison with grade III tumors (results not shown). Subcutaneous MCTs (n = 5) were diagnosed as low grade (n = 4) and high grade (n = 1). There were too few dogs with subcutaneous MCTs to be statistically compared with those with cutaneous tumors.
Histologic grade was a significant factor for clinical outcome (Table 2). High-grade MCTs had significantly decreased DFI and decreased time to local recurrence and metastasis than did lower-grade MCTs. For survival, grade could not be statistically measured, as all MCT-related deaths were due to high-grade tumors (ie, 100% were in 1 category). The small number of cases precluded statistical measurement of the effect of grade for distant occurrences.
MI was categorized into groups per cutoff values previously published for cutaneous (MI = 7) 22 and subcutaneous (MI = 4) 37 MCTs (Table 3). Results were the same either way, as these data had no MCTs with MI >4 or ≤7. The majority of tumors had a low MI. Tumors with a MI >7 resulted in a higher proportion of MCT death, local recurrence, and metastasis. MI was a significant factor for decreased survival time, DFI, time to local recurrence, and metastasis (Table 2).
Risk Factor Expression for 22 Dogs With Cutaneous and Subcutaneous MCTs.a
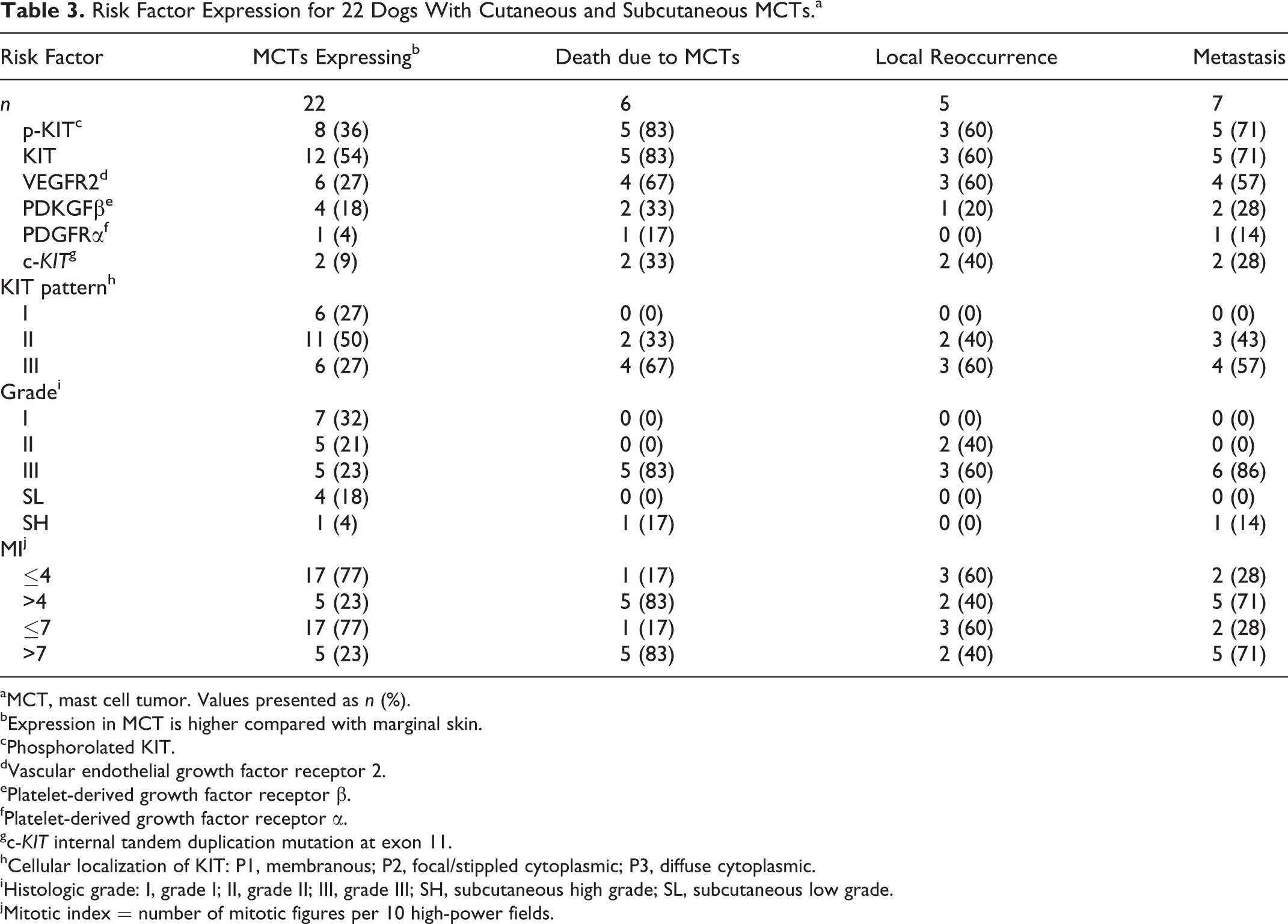
aMCT, mast cell tumor. Values presented as n (%).
bExpression in MCT is higher compared with marginal skin.
cPhosphorolated KIT.
dVascular endothelial growth factor receptor 2.
ePlatelet-derived growth factor receptor β.
fPlatelet-derived growth factor receptor α.
gc-KIT internal tandem duplication mutation at exon 11.
hCellular localization of KIT: P1, membranous; P2, focal/stippled cytoplasmic; P3, diffuse cytoplasmic.
iHistologic grade: I, grade I; II, grade II; III, grade III; SH, subcutaneous high grade; SL, subcutaneous low grade.
jMitotic index = number of mitotic figures per 10 high-power fields.
KIT Cellular Localization Patterns and c-KIT Mutational Analysis
KIT cellular localization (Figs. 4–6) was determined for all cases. KIT pattern distribution for all dogs was as follows: membranous/pattern I (n = 6), focal/stippled cytoplasmic/pattern II (n = 14), and diffuse cytoplasmic/pattern III (n = 7). For subcutaneous MCTs, diffuse KIT pattern expression was present only in the high-grade tumor. Membranous and focal/stippled groups were combined for statistical analyses, as there was no statistical difference between these (results not shown). Diffuse cytoplasmic KIT pattern was a significant risk factor for decreased survival time, DFI, rate of local recurrence, and metastasis (Table 2). Diffuse KIT expression was demonstrated in 7 of the 10 MCTs from dogs that died of metastasis and 4 of the 7 MCTs from dogs that developed local recurrence.
c-KIT mutations were detected in 7 MCTs from 5 dogs with cutaneous MCTs (Fig. 7). All were grade III MCTs. No c-KIT mutations were found in subcutaneous MCTs. For the 2 dogs where the original and locally recurring MCT was available, identical mutations were present in both the original and locally recurring MCTs, consisting of 45 base pairs (bp) and 50-bp ITD mutations in exon 11 of c-KIT, respectively. One MCT, a grade III tumor removed from the neck of a 7-year-old Sharpei, had a 43-bp ITD mutation in intron 11, and the remainder of detectable c-KIT mutations were ITD mutations within exon 11 of KIT. Three of the 5 dogs with c-KIT mutations developed local recurrence, and all 5 dogs died from metastatic disease. Follow-up times for the 5 dogs with c-KIT mutations were 31, 43, 78, 194, and 708 days; the 2 dogs with the longest survival time were treated with chemotherapy and toceranib. Insufficient cases with c-KIT mutations were available for survival analysis of this parameter.
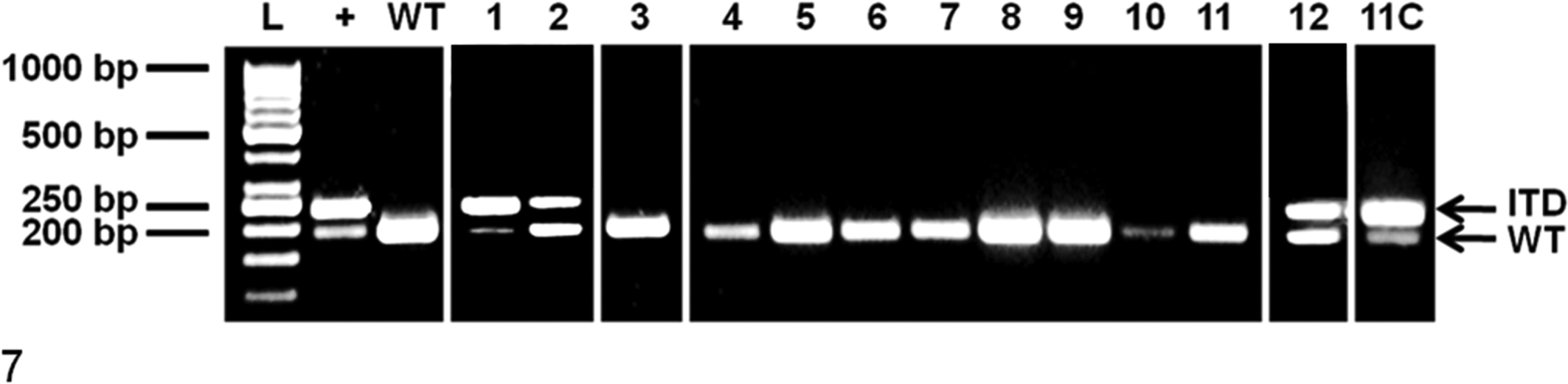
Polymerase chain reaction amplification of exon and intron 11 of c-KIT from canine cutaneous and subcutaneous mast cell tumors. +, positive control (mast cell tumor continuous cell line) heterozygous for wild-type allele (190 base pairs [bp]) and mutant allele (238 bp); 1–12, mast cell tumor number; 11C, cultured cells derived from sample 11; ITD, internal tandem duplication (238 bp); L, 50-bp ladder; WT, canine blood homozygous for wild-type allele (190 bp).
RTK Expression in MCT Tissue and Marginal Skin
Representative Western blots for phosphorylated KIT (p-KIT), KIT, VEGFR2, PDGFRα, and PDGFRβ expression and phosphorylation for MCTs and marginal skin are shown in Figures 8 and 9. Mast cell tumors had heterogeneous expression profiles for p-KIT, KIT, VEGFR2, and PDGFRβ, and variable levels of these markers were detectable in marginal skin. Total KIT was expressed in canine cerebellum and with variable intensity in MCTs but was not detectable in marginal skin (Fig. 8). The majority of tumors (16 of 22) expressed KIT, but 4 of these demonstrated very weak levels, which were detected only after prolonged exposure times (2 hours; results not shown). These 4 cases were considered negative for KIT expression for subsequent analysis.
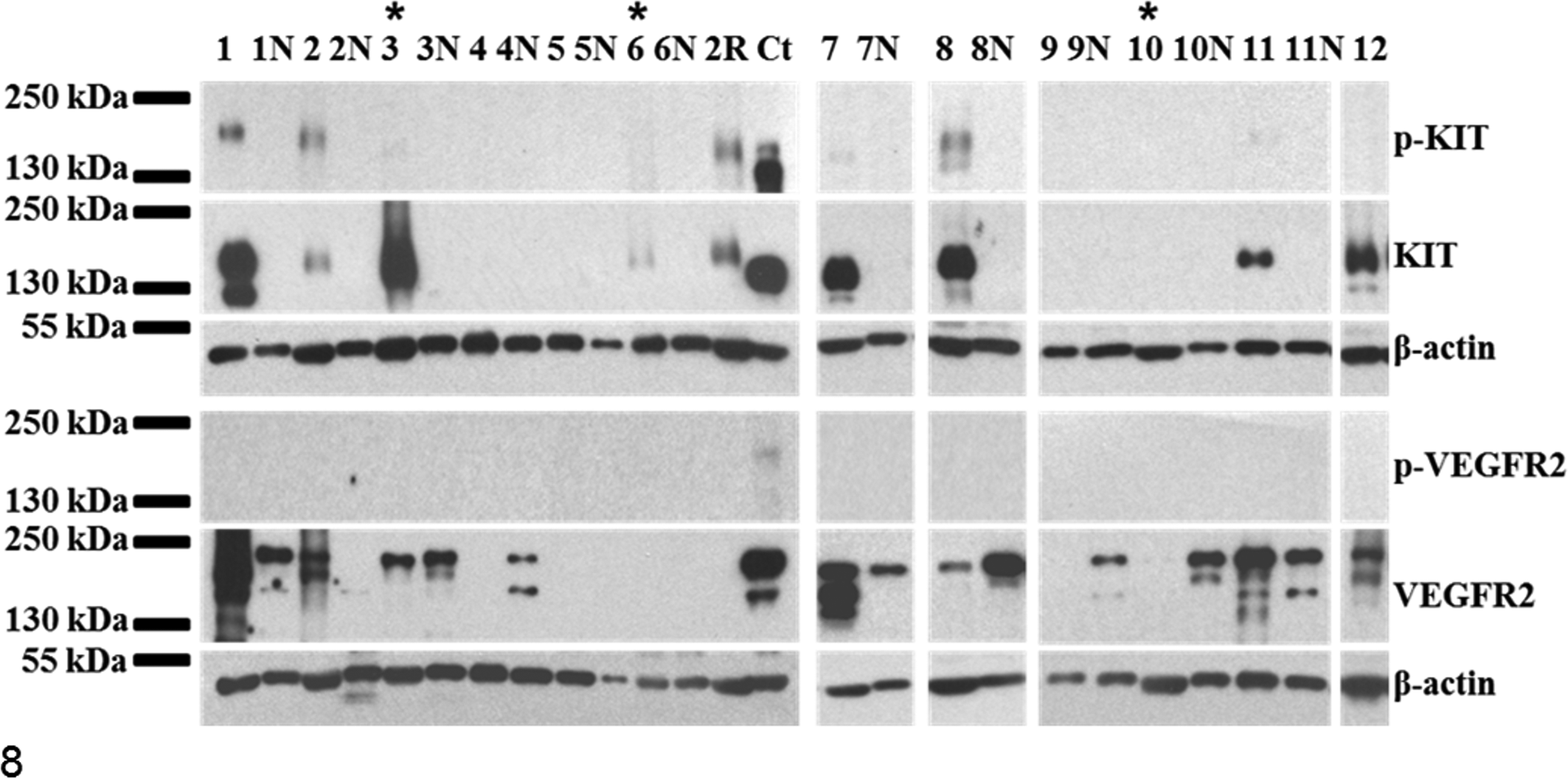
Receptor tyrosine kinase expression profiles for canine cutaneous and subcutaneous mast cell tumors (MCTs). Representative Western blots of phosphorylated KIT (p-KIT) and total KIT, as well as phosphorylated vascular endothelial growth factor receptor 2 (p-VEGFR2) and total VEGFR2, from MCTs, marginal skin (N), and canine controls (Ct; canine cerebellum [KIT] or canine aorta [VEGFR2]). Subcutaneous MCTs are indicated with asterisks (sample Nos. 3, 6, 10). Total and/or p-KIT is seen in canine cerebellum and with variable intensity in some MCTs (sample Nos. 1–3, 6–8, 11). Expression of VEGFR2 is detected in canine marginal skin as well as MCTs with variable intensity. VEGFR2 expression is more strongly expressed in MCTs relative to marginal skin in sample Nos. 1, 2, 7, 11. Phosphorylation of VEGFR2 is demonstrated in canine aorta only. β-actin was used as a loading control.
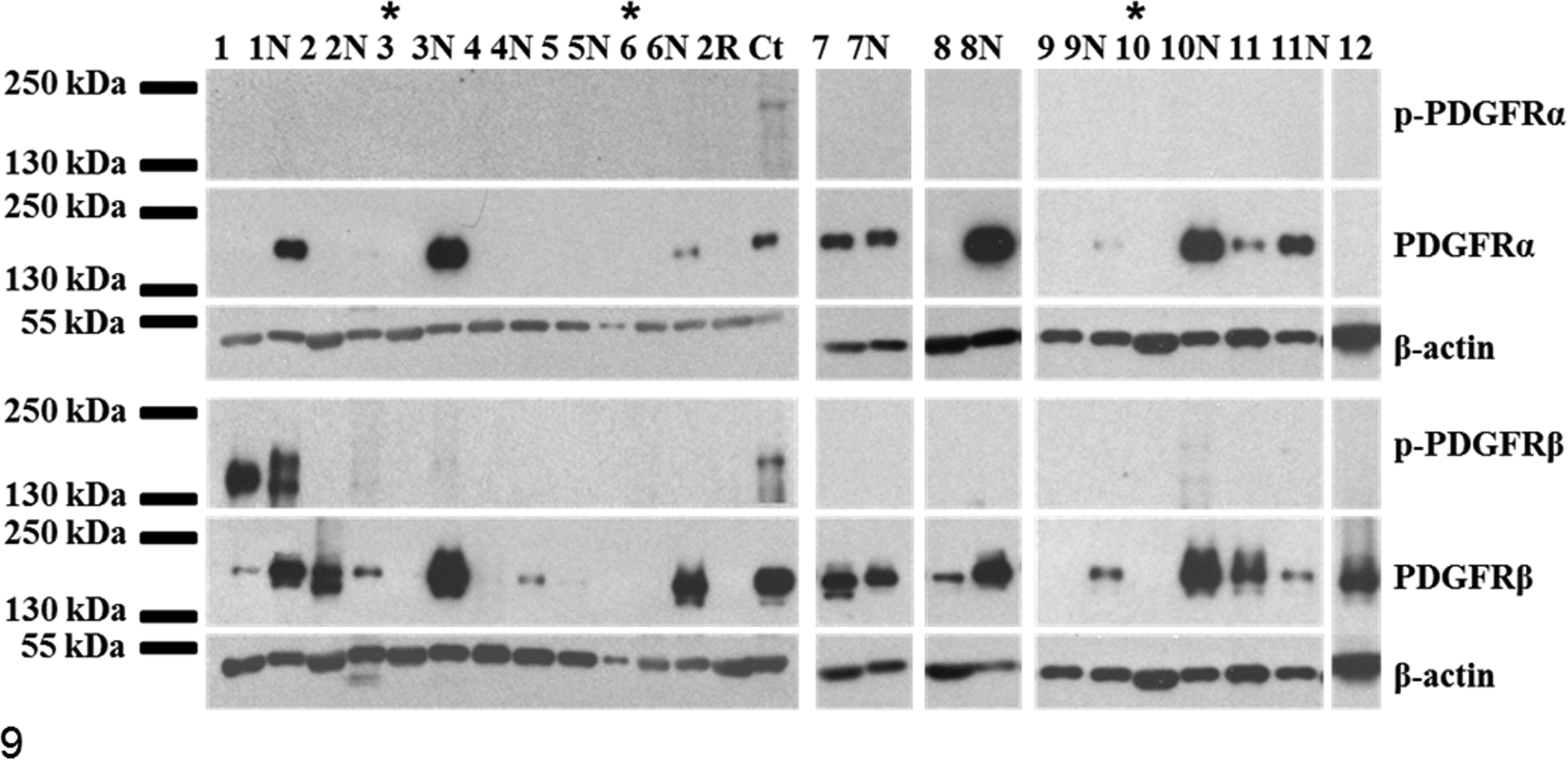
Receptor tyrosine kinase expression profiles for canine cutaneous and subcutaneous mast cell tumors (MCTs). Representative Western blots of phosphorylated platelet-derived growth factor receptors α and β (p-PDGFRα and p-PDGFRβ) and total PDGFRα and PDGFRβ from MCTs, marginal skin (N), and canine controls (Ct; canine aorta). Subcutaneous MCTs are indicated with asterisks (sample Nos. 3, 6, 10). Expression of PDGFRs is detected in canine marginal skin as well as MCTs with variable intensity. PDGFRβ expression is comparatively increased over marginal skin in 3 cases (sample Nos. 2, 7, 11). Phosphorylated PDGFRβ is strongly expressed in MCT sample No. 1 and its corresponding marginal skin and in aorta and weakly expressed in marginal skin from sample Nos. 2, 3, and 10. PDGFRα expression is absent in the majority of MCTs (except case Nos. 7, 11) but strongly expressed in the majority of marginal skin samples (1N, 3N, 6N, 8N, 10N). Phosphorylation of PDGFRα is demonstrated in canine aorta only. β-actin was used as a loading control.
KIT was phosphorylated in 10 MCTs (representative examples in Fig. 8), 2 of which were subcutaneous. All demonstrated strong total KIT expression; however, relative phosphorylation was considerably greater in some cases (eg, sample Nos. 2 and 2R; Fig. 8), which indicated increased cellular activity. Both MCTs from this dog possessed a 50-bp ITD mutation in exon 11 of c-KIT, and this dog died from metastatic disease despite aggressive treatment with chemotherapy and toceranib.
In general, phosphorylated KIT was detectable in dogs with unfavorable clinical outcomes, as were those with diffuse cytoplasmic KIT expression (Table 3). Phosphorylated KIT expression was a statistically significant risk factor for decreased survival time, DFI, and increased rate of metastasis (Table 2). Of the 8 dogs with MCTs having p-KIT expression, 5 died from MCTs. The median survival time for dogs with MCTs having p-KIT expression was 270 days (125 to 708 days) as compared with those without detectable p-KIT (median survival time not reached; 89% of dogs alive at 421 days; Fig. 10). Local recurrence and metastasis were seen in 38% (3 of 8) and 62% (5 of 8) of these dogs, respectively. Total KIT overexpression, compared with marginal skin, was not a significant risk factor for any outcome.
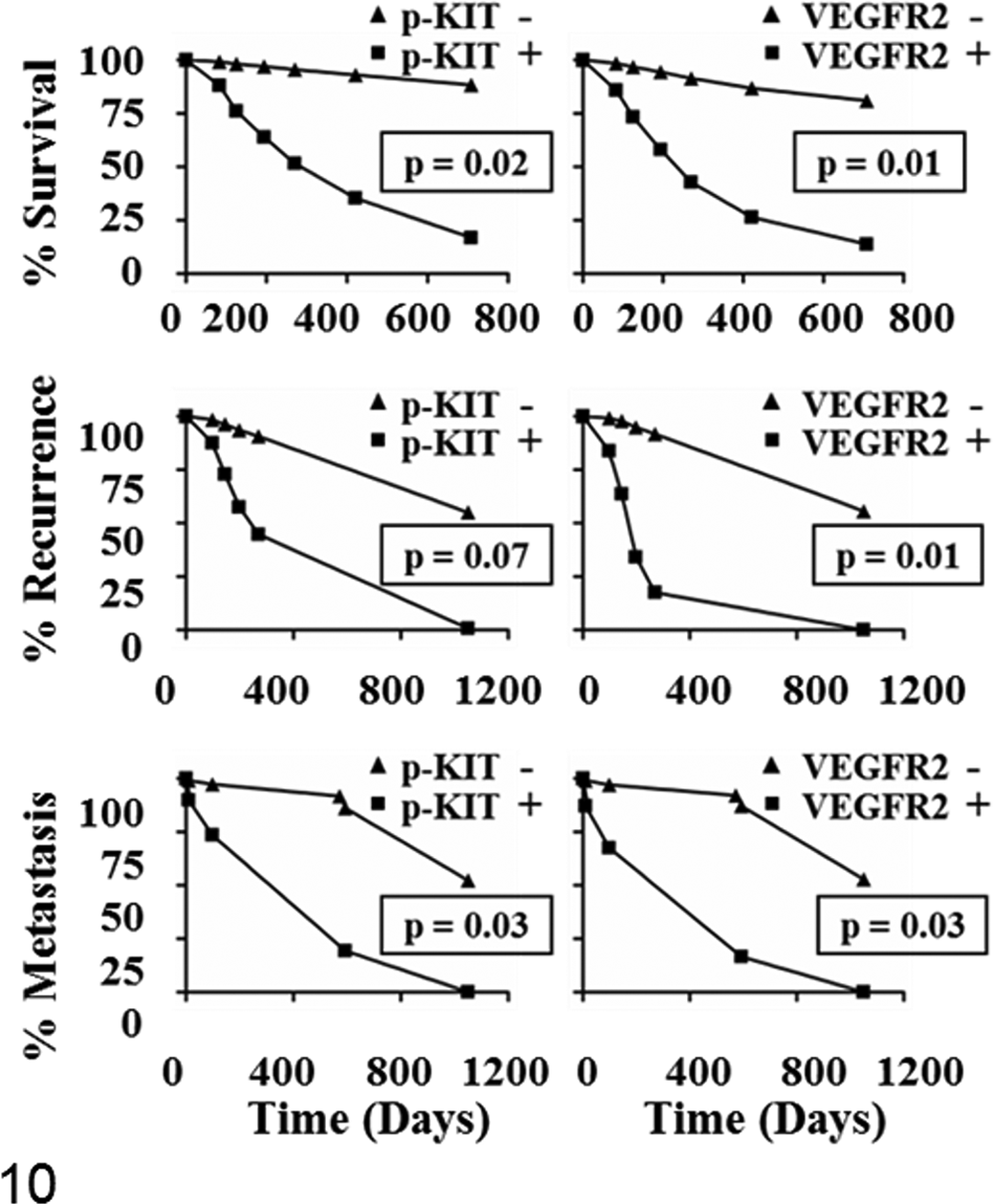
Predicted survival curves for dogs with cutaneous and subcutaneous mast cell tumors (MCTs). Outcomes were compared per the presence or absence of phosphorylated KIT (p-KIT) and vascular endothelial growth factor receptor 2 (VEGFR2) expression in MCTs. Cases were considered positive for expression if levels were higher in MCTs than marginal skin, which served as a control. Curves are based on univariable Cox proportional hazard models. Survival time and time to metastasis for dogs assessed as positive for p-KIT (left) and VEGFR2 (right) were significantly decreased as compared with those not overexpressing the risk factors. The rate of local recurrence was significantly increased for dogs having tumors with greater VEGFR2 expression (right, middle) than marginal skin.
VEGFR2 Expression and Phosphorylation in MCT Tissue Lysates
VEGFR2 was expressed in MCTs and marginal skin (Fig. 8). Cases that had greater relative expression in tumors versus marginal skin were considered positive for analytic purposes (Table 3). Of those dogs, 67% (4 of 6) died from MCTs. Phosphorylated VEGFR2 was not detected in tissue lysates (Fig. 8). Median survival time, time to local recurrence, and time to metastasis were 270 days (98–270 days), 145 days (98–270 days), and 270 days (98–270 days), respectively, for dogs with MCTs with relatively greater VEGFR2 expression than marginal skin (Fig. 10). VEGFR2 expression was a statistically significant risk factor for decreased survival time, DFI, local recurrence, and increased rate of metastasis (Table 2).
PDGFRα and PDGFRβ Expression and Phosphorylation in MCT Tissue Lysates
PDGFRβ expression was detected in both tumor and marginal skin (Fig. 9) and was expressed with greater intensity in MCT tissue (vs marginal skin) in 4 cases. Phosphorylation of PDGFRβ was seen in only 1 case (sample No. 1); however, the significance of this is uncertain, as phosphorylation was also seen in normal marginal skin samples. Three of the 5 subcutaneous MCTs had PDGFRβ expression, but this was not greater than marginal skin.
PDGFRα was detected in some skin samples from surgical margins (n = 14), but this was demonstrated in only 4 MCTs. PDGFRα was overexpressed relative to marginal skin in 1 case (sample 10; Fig. 9). As there were so few tumors that expressed PDGFRα, this was not assessed as a risk factor. Relative overexpression of PDGFRβ was not determined to be a significant risk factor for any clinical outcome (Table 2).
Statistical Correlations Among Diagnostic Markers
Several diagnostic markers were highly positively correlated (ie, >50%; Table 4). The most highly correlated variables were p-KIT and KIT (r = 0.69; P < .001). Phosphorylated KIT overexpression (vs marginal skin) was also significantly correlated with high VEGFR2 expression (r = 0.60; P = .01), MI (r = 0.49; P = .04), and high histologic grade (r = 0.59; P < .01). Diffuse cytoplasmic KIT localization was significantly correlated with histologic grade (r = 0.64; P = .01), MI (r = 0.48; P = .05), c-KIT mutations (r = 0.58; P = .04), and KIT overexpression (r = 0.49; P = .03). PDGFRβ was not significantly associated with any other marker except VEGFR2 (r = 0.51; P = .04). VEGFR2 expression was significantly correlated with histologic grade (r = 0.54; P = .02) but not MI.
Associations Between Risk Factors for Canine Cutaneous and Subcutaneous Mast Cell Tumors.a
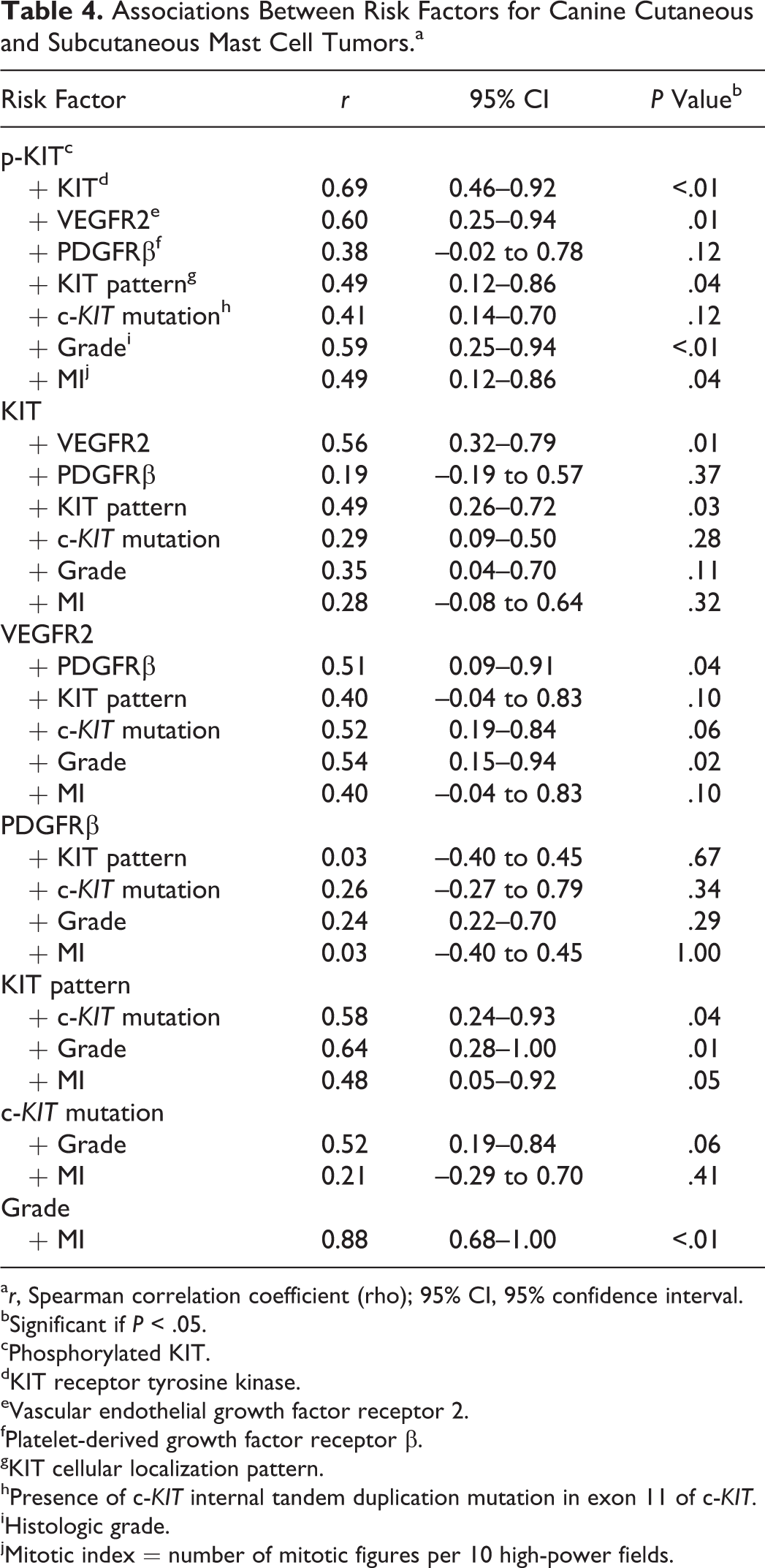
a r, Spearman correlation coefficient (rho); 95% CI, 95% confidence interval.
bSignificant if P < .05.
cPhosphorylated KIT.
dKIT receptor tyrosine kinase.
eVascular endothelial growth factor receptor 2.
fPlatelet-derived growth factor receptor β.
gKIT cellular localization pattern.
hPresence of c-KIT internal tandem duplication mutation in exon 11 of c-KIT.
iHistologic grade.
jMitotic index = number of mitotic figures per 10 high-power fields.
RTK Expression and Activation in Cultured Neoplastic Mast Cells
Cells from 10 MCTs were isolated for in vitro culture. Purity and viability were >90% as assessed by cytology of cytospins (Fig. 3) and Trypan blue exclusion. KIT, VEGFR2, and PDGFRβ were shown to be variably expressed in cells as assessed by Western blotting (Fig. 11). RTK expression and phosphorylation were generally more readily detectable in cells than in tissue lysates.
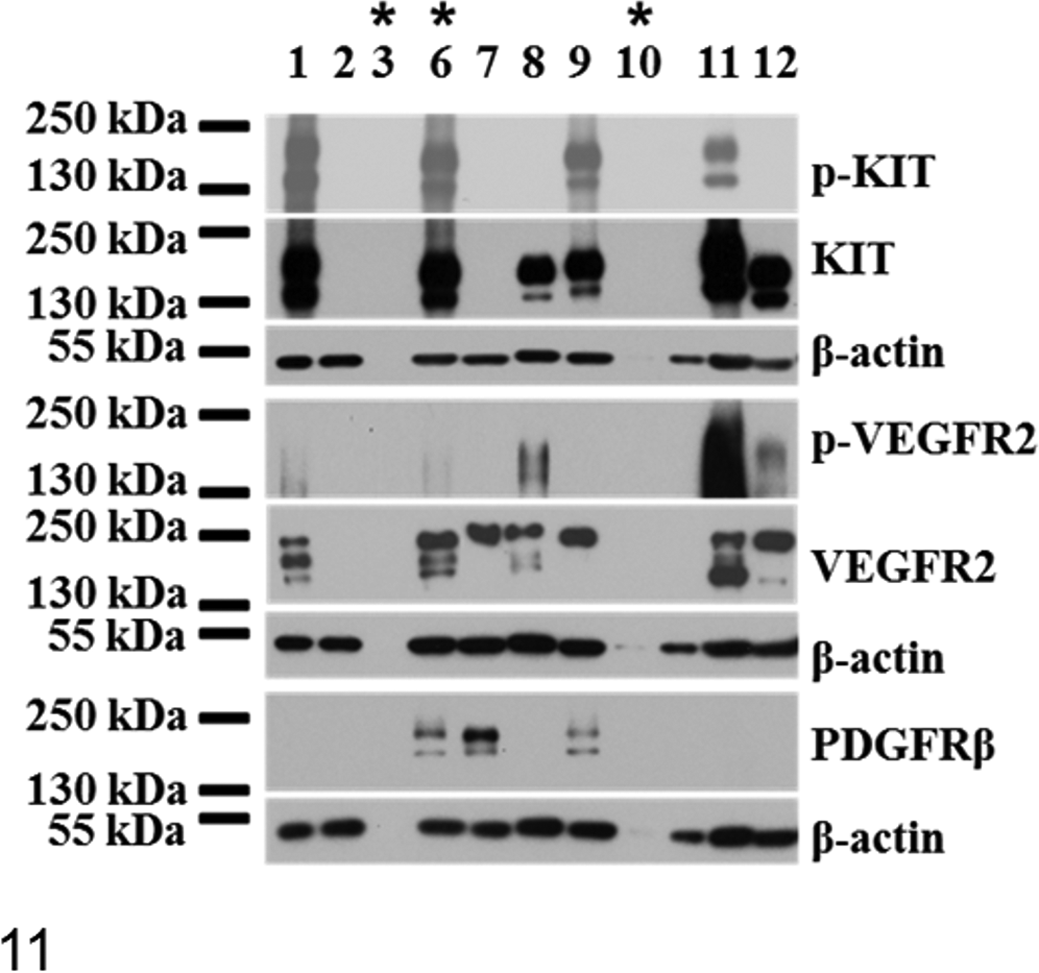
Receptor tyrosine kinase expression profiles of cultured cells derived from cutaneous and subcutaneous canine mast cell tumors. Western blots of phosphorylated KIT (p-KIT) and total KIT, phosphorylated vascular endothelial growth factor receptor 2 (p-VEGFR2) and total VEGFR2, and total platelet-derived growth factor receptor β (PDGFRβ) from MCT lysates of serum-starved cells cultured from mast cell tumors. Subcutaneous MCTs are indicated with asterisks (sample Nos. 3, 6, 10). All cells expressed detectable KIT with prolonged exposures (2 hours, not shown), but phosphorylation was detected in only some MCTs (sample Nos. 1, 6, 9, 11). Both high molecular weight (145 kDa; putative glycosylated) and lower molecular weight (130 kDa; putative nonglycosylated) isoforms of KIT are seen in cell lysates; however, the 130-kDa band is much less intense in some cells (sample Nos. 8, 9). VEGFR2 is detected in most cells, and phosphorylated VEGFR2 (Tyr 951) is seen in approximately half of samples. Cells show variable isoform expression, presumably corresponding to the glycosylated (230 kDa), partially glycosylated (210 kDa), and nonglycosylated (150 kDa) forms reported for human cells. Detectable PDGFRβ is expressed in 3 samples (Nos. 6, 7, 9).
For KIT, the molecular weight for bands was higher in some cells (eg, sample Nos. 1, 11; Fig. 11) than others (eg, sample No. 12; Fig. 11), and this was also seen in some MCT tissue correlates (Fig. 8). One of these had a demonstrable c-KIT ITD mutation (sample No. 1), which may explain the larger receptor size in that case.
VEGFR2 was expressed in most cells (Fig. 11), and unlike in tissues, phosphorylation was detected in 50% of samples (5 of 10), with relative phosphorylation highest for sample No. 11, indicating greater VEGFR2 activation. With the exception of the dog that died from unknown causes (sample No. 11), all dogs that had MCTs demonstrating phosphorylated VEGFR2 in neoplastic cells died from MCTs.
For 2 cases (samples Nos. 6, 9; Fig. 11), VEGFR2 and PDGFRβ expression was detected in cells but not in tissue correlates (Figs. 8, 9). Conversely, RTK total expression detected in tumors was occasionally lacking in isolated MCT cells. For instance, PDGFRβ was detected in tissue lysate for only sample No. 1, and neither VEGFR2 nor PDGFRβ was detectable in cells but was detectable in tissue for sample No. 2 (Figs. 8, 9, 11); thus, the expression of VEGFR2 and PDGFRβ in these MCTs was mainly due to the stroma. For sample No. 12, PDGFRβ was observed only in tissue lysate, indicating stromal expression as well. PDGFRβ was expressed in isolated MCT cells from some cases (Fig. 11), but this was detectable in tissue correlates in only 1 case (sample No. 7; Fig. 9). Levels of total PDGFRα were not detectable in isolated neoplastic cells, even for the 2 MCTs that showed tissue expression (sample Nos. 7, 11); thus, this was likely predominantly expressed by stromal components. Phosphorylation of PDGFR was undetectable in cell lysates.
Discussion
In this study, canine MCTs heterogeneously expressed KIT, VEGFR2, and PDGFRβ in tumor tissue and cultured neoplastic cells. Expression profiles among individual dogs vary. Phosphorylated KIT, native KIT, and VEGFR2 were useful prognostic markers and may prove useful in selecting patients amenable to RTK inhibition. These data therefore have implications for the development of treatment protocols based on the selection of specific receptor inhibitors.
Phosphorylated KIT was detected in both c-KIT-mutated and nonmutated MCTs in dogs with poor outcomes in this study. This finding has been reported, 33 but the present study is the first to show that p-KIT is associated with poorer clinical outcomes. The presence of a c-KIT mutation is associated with high-grade cutaneous MCTs, 43 and detection of these mutations identifies those dogs that will potentially respond well to RTK inhibition. 27 The results of this study support these findings and show that the presence of p-KIT may identify dogs that could also benefit from RTK inhibition. The role of c-KIT mutations in subcutaneous MCTs is not known and is yet to be detected. 37 Activated KIT is linked to c-KIT mutations, as this leads to conformational changes in the receptor that permits constitutive signaling. 5 The detection of activated KIT without exon 11 mutations suggests that other causative mechanisms are responsible. These MCTs may have other c-KIT mutations, as in exon 8 or 9, as has been reported for some canine MCTs. 23 There are several alternative possibilities, including autocrine and paracrine stem cell factor production, spontaneous homodimerization, heterodimerization with other RTKs, loss of phosphatase function, or failure of degradation processes such as ubiquitinization. 3,15,35 Diffuse cytoplasmic KIT localization is thought to be due to aberrant receptor trafficking or degradation 35,39 and is a useful predictor of poor outcome in cutaneous 39 and subcutaneous MCTs. 37 This present study supported those findings and, in addition, demonstrated that p-KIT expression compared similarly to KIT localization as a prognostic test. It is not known if KIT activation and aberrant localization are directly linked, but these data showed significant correlation of these markers. Presence of p-KIT is a promising marker for aggressive MCT disease, and it could be used to distinguish mechanisms of KIT dysregulation in future studies. In a few cases, Western blotting of tumor tissue lysate was not concordant with immunohistochemical detection of KIT. The reasons for this are not clear but could include (1) differences in antibody affinity between the reagents used for immunohistochemical versus Western blotting and (2) heterogeneity of the tumor regions used for each analysis.
This is the first study to demonstrate the potential utility of VEGFR2 expression or activation with clinical outcomes in canine MCTs. Strong VEGFR2 expression was detected in MCT biopsies and neoplastic cell lysates, suggesting that neoplastic cells and the tumor stroma are potential targets of RTK inhibitors. In this study, VEGFR2 expression was a significant risk factor for decreased survival, DFI, and time to local recurrence and metastasis. The detection of activated receptors in cancer cells indicates that MCTs could be amenable to anti-VEGF therapy, targeting a potential autocrine loop.
It is likely that tumor cells and stroma play important and potentially synergistic roles in MCT progression; both are implicated in the progression of other canine neoplasms. 1,9,28,31,42 Increased VEGF and VEGFR2 expression is reported in canine cancers such as lymphoma, mammary carcinoma, vascular tumors, 1,28,42 and recently MCTs. 31,34 One group reported VEGFR2 receptors and VEGF within MCT cells and found that VEGF expression was subjectively more intense in higher-grade MCTs as assessed with immunohistochemistry. 34 Additionally, that study demonstrated VEGF production in conditioned media produced by neoplastic cells, which suggests that autocrine VEGFR2 activation may be a mechanism for tumor progression or neoplastic cell survival. 34 VEGFR2 phosphorylation was not determined to assess this process. That study used a single cell line, the C2 cell line, which is known to possess activating c-KIT mutations 24 ; thus, it may not have relied on VEGF signaling pathways. In this present study, Western blots of cell lysates showed that VEGFR2 expression is not uniformly present within MCT cells and, more important, constitutive phosphorylation was detected in only 50% of cell lysates (5 of 10), indicating that VEGFR2 activation is heterogeneous. Several cell lines would be needed to understand the role of this receptor in MCTs.
PDGFRβ was expressed in many aggressive MCTs, from neoplastic cells, stromal components within the tumor, or both. Expression of PDGFRβ was demonstrated in the MCT stroma, in marginal skin, and in some neoplastic cells, while PDGFRα was predominantly found in marginal skin. In the majority of cases, PDGFRβ and PDGFRα expression was greater in marginal skin than MCTs, suggesting that overall expression is reduced by effacement of stromal tissue by (nonreceptor bearing) neoplastic cells or that expression of these receptors is lost in some neoplastic mast cells. Three cases strongly expressed PDGFRβ in cell lysates; thus, this receptor is expressed heterogeneously in neoplastic cells. PDGFRβ overexpression in tumor lysates (vs marginal skin) was not found to be a statistically significant risk factor for any clinical outcome. This may be due to the small number of cases.
As PDGFR signaling has wide implications for tumor progression in humans 7,8,16,32 and are implicated in the progression of animal neoplasms, 6,21 this should be further explored in canine MCTs.
This study examined a relatively small number of cases. This influenced statistical analyses, as there were wide confidence intervals. For this reason, subcutaneous MCTs could not be compared with cutaneous ones to further differentiate these variants. Risk factors could also not be analyzed with multivariable statistics, so the effects of confounding factors and interactions could not be examined. Additional factors that potentially influenced the data were a lack of definitive confirmation of metastasis, concurrent neoplasia other than MCTs (eg, osteosarcoma), and varying treatments. Furthermore, 3 cases were excluded from statistical analysis, as no marginal skin was available. Two of these had c-KIT mutations; thus, the study may have underestimated the significance of c-KIT mutations for prognosis.
For statistical purposes, tumors were classified as positive for VEGFR2 or PDGFR if expression of either receptor was increased relative to skin at the surgical margin. This subtracted background stromal effects, which likely vary among individuals, anatomic locations, and physiologic states. This selection may be too simplistic. Early tumor progression may lead to diminished expression of these RTKs, due to effacement or displacement of stromal components by (nonreceptor expressing) neoplastic cells. At later stages, neoplastic cells may acquire RTK expression, or stromal components (eg, blood vessels and fibroblasts), which express these receptors, may predominate within certain tumors. Both these events may confer malignancy and were not distinguishable in this study. There may have been stromal component variation among marginal skin samples (eg, amount of adipose tissue) or as a result of different therapy, including prior surgery. Mast cells are very difficult to sustain in vitro, 2 so successful short-term culture of only 10 MCTs was possible. Expression levels of cells and tissue lysates could not be compared for all cases.
Despite these limitations, this study shows that canine MCTs have a spectrum of RTK expression and that individual profiles vary. This may explain variable clinical responses to treatment with toceranib, an RTK inhibitor. 26,27 Although KIT is expressed only by neoplastic cells, VEGFR2 and PDGFRβ are expressed in neoplastic cells and stroma. RTK inhibitor therapy targets neoplastic cells and supporting tissue. Because some neoplastic cells demonstrate activated VEGFR2, at least some MCT cells may depend on VEGF signaling, and this should be explored at a cellular level.
Footnotes
Acknowledgements
We thank the veterinary clinics who participated in this study for providing follow-up information for dogs. We acknowledge both the Ontario Veterinary College Pet Trust Fund and the Rouse Family Foundation for funding this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors received financial support from the OVC Pet Trust Fund.