
Abstract
The microbiome is the complex collection of microorganisms, their genes, and their metabolites, colonizing the human and animal mucosal surfaces, digestive tract, and skin. It is now well known that the microbiome interacts with its host, assisting in digestion and detoxification, supporting immunity, protecting against pathogens, and maintaining health. Studies published to date have demonstrated that healthy individuals are often colonized with different microbiomes than those with disease involving various organ systems. This review covers a brief history of the development of the microbiome field, the main objectives of the Human Microbiome Project, and the most common microbiomes inhabiting the human respiratory tract, companion animal digestive tract, and skin in humans and companion animals. The main changes in the microbiomes in patients with pulmonary, gastrointestinal, and cutaneous lesions are described.
The various microorganisms (fungi, protozoa, bacteria, archaea, bacteriophages, and viruses of eukaryotes) that live in and on the bodies of humans and other animals are more than a simple collection of microbes. The microbiome encompasses the full complement of microorganisms, their genes, and their metabolites. The microbiome has co-evolved with humans and animals, thereby assisting in digestion and detoxification, supporting immunity, protecting against invading pathogens, and maintaining overall health. At 1014 species, comprising at least 20 million unique microbial genes, the microbiome constitutes the largest genetic component of the human and animal superorganism. Microbial ecologists who studied microorganisms and microbial communities in the environment recognized early on that most microorganisms in nature were not readily culturable and so developed alternate approaches to the study of microbial communities. An early and broadly adopted approach for investigating microorganisms in the environment was the use of the 16 S ribosomal RNA (rRNA) gene as a taxonomic marker for interrogating bacterial diversity in nature. 71 With the growth of non–culture-based molecular techniques to study environmental microorganisms and communities, some medical microbiologists started using these tools to study the human body and found far greater microbial diversity than expected, even in well-studied sites such as the oral cavity. 7,15,70
In the infectious diseases field, recognition was growing that many diseases could not satisfy Koch’s postulates as their pathogenesis appeared to involve multiple microorganisms. The term polymicrobial diseases was coined to describe these diseases resulting from concurrent infection with multiple infectious agents, 11 as seen with abscesses, AIDS-related opportunistic infections, conjunctivitis, gastroenteritis, hepatitis, otitis media, periodontal diseases, respiratory diseases, and genital infections. However, we now recognize added complexity in these diseases, as we see these as entire microbial communities and begin to understand how the bacteria that comprise them interact with each other and with the host. In an essay on the history of microbiology and infectious disease, Lederberg 73 coined the term microbiome and called for “a more ecologically-informed metaphor” to understand the relationship between humans and microbes.
This review includes an introduction to the National Institutes of Health (NIH) Human Microbiome Project (HMP) and considers the microbiota inhabiting the human respiratory tract, the digestive tract in companion animals, and the skin in humans and dogs in health and disease.
The NIH Human Microbiome Project
In order to better evaluate the role of the microbiome in health and disease, the NIH launched the HMP, which focused on surveying microbiomes present in different organ systems (www.commonfund.nih.gov/hmp). The NIH-HMP was divided into 2 phases: Phase 1 surveyed the microbiomes of major body regions in healthy individuals and in those with disease, and Phase 2 focused on the biological properties of these microbiomes. The initial studies focused on the digestive tract 23,39 and demonstrated the tremendous complexity as well as the functional potential of the human microbiome.
Phase 1 Healthy Adult Cohort Study
During the first phase of HMP (2007–2012), the major objectives were to (1) survey the microbiomes of healthy adults to produce a reference data set, (2) develop a catalog of genome sequences of microbial reference strains, and (3) evaluate the properties of microbiomes associated with specific diseases in a collection of HMP Demonstration Projects (Table 1). 96 The microbiomes were surveyed in 5 major body regions of healthy adults (airway, skin, oral cavity, digestive tract, and vagina). From these studies, the HMP Consortium published 2 landmark papers in 2012 describing the range of normal microbial variation among healthy adults in a Western population. 49,50 One major finding was that even though microbial community structure varied greatly between body habitats, the potential metabolic capabilities encoded in these communities’ metagenomes were much more constant. That is, although microbial taxonomic composition varied among healthy individuals, their collective metabolic functions remained remarkably stable within each body site. 1
Major objectives of the Human Microbiome Project divided into 2 phases.

Another key resource from this phase was the HMP reference microbial genome sequence catalog, which includes the largest collection of human-associated microbial genomes sequences, including bacteria, bacteriophages, viruses of eukaryotes, and microbial eukaryotes (NCBI, http://www.ncbi.nlm.nih.gov/bioproject/43021; Bioproject PRJNA28331). Nonetheless, with tens of thousands of bacterial strains and unknown numbers of fungal, viral, and protists in the human microbiome, much work remains to create a complete catalog of reference genomes. 31 Approximately 15% of the samples collected in the healthy cohort study were sequenced by whole genome shotgun (WGS) sequencing to produce metagenomic sequences for the 5 major body regions of the study.
To evaluate and compare the healthy cohort study with other individuals, 15 Demonstration Project studies were launched focusing on gastrointestinal diseases (Crohn’s disease, ulcerative colitis, pediatric inflammatory bowel syndrome, neonatal necrotizing enterocolitis, esophageal adenocarcinoma, and idiopathic pediatric fever), urogenital conditions (bacterial vaginosis, men with circumcision), skin diseases (eczema, psoriasis, and acne), and patients with obesity.
The outcomes from Phase 1 accomplished many things for the field though it also raised new questions about the best approaches for identifying characteristic signature phenotypes. It was noted that in many cases, taxonomic composition of the microbiomes alone was not sufficient to define a core microbiome associated with specific diseases or states of health. Rather, predicted metabolic pathways of these microbiomes appeared to move us closer to this goal.
Phase 2 of the HMP (2013–2015)
Phase 2 of the HMP (2013–2015) focused on developing a data set of multiple biological properties of the microbiome and host from well-characterized cohort studies (www.hmp2.org). A cardinal property of microbes is their versatile metabolic capabilities, which are not taxon specific. In other words, more than 2 unrelated microbes may have the ability to break down a common component in our diet, and this can mean that microbial community makeup may not be the best biomarker property to characterize specific human conditions. Therefore, Phase 2 was designed to collect multi-omic biological properties of the microbiome, such as the gene expression profiles (ie, the metatranscriptome), 33,48,128 protein profiles (ie, the metaproteome), 127 profiles of metabolites from host and microbome (ie, the metabolome), 16 and related relevant host properties from well-characterized cohorts. These studies are focusing on the microbiome and host (mother and child) for pregnant women at risk for preterm birth, the gut microbiomes of cohorts at risk for inflammatory bowel disease (IBD), and the gut and nasal microbiomes from cohorts at risk for type 2 diabetes. 54
These data and the resultant integrated data set, which will be deposited in public databases, will allow the scientific community to evaluate microbiome properties or combinations of properties and provide new insights into the role of the microbiome in health and in disease.
The Respiratory Tract Microbiome
The Upper Respiratory Tract Microbiome
The human upper respiratory tract (URT), including the nose, throat, and oral cavities, is colonized by a complex and dynamic microbial community. Collectively, the upper airways, in particular the oral cavity, represents the most diverse microbiome site in the body, even harboring more bacterial species richness than the digestive tract. 51,75,111 As the site of initial interactions with many environmental microbes through breathing and ingestion, an important role of the commensal microbiota is the first line of defense against pathogens by outcompeting potential colonizing pathogens (microbial antagonism). 65,82 Essentially, by occupying all binding sites in the URT, any invading pathogen must somehow contend with these organisms, in addition to the host defenses. Importantly, the microbiota of the airways, like those at other mucosal surfaces, are integral in priming and educating the immune system 72 and regulating immunity in the lung in response to infection. 52 To date, most of the research has used 16 S rRNA gene sequencing to characterize the bacterial communities of the respiratory tract. Fungal and viral communities have not been studied, with the exception of pathogen-focused studies.
The nasal passages and oropharynx harbor a distinct microbiota. In healthy adults, the nasal passages are typically dominated by Actinobacteria (Propionibacterium, Corynebacterium) and Firmicutes (Staphylococcus), whereas Firmicutes (Veillonella, Streptococcus, Staphylococcus) are more prevalent in the oropharynx (Fig. 1). 14,51,74 Importantly, the resident microbiota of the upper airways includes many pathogenic bacteria that colonize asymptomatically such as Streptococcus pneumoniae, Staphylococcus aureus, Moraxella catarrhalis, and Haemophilus influenzae. 35 For these organisms, the distinction between commensal and opportunistic pathogen is blurred. When host defense at these sites and in the lower respiratory tract (LRT) are compromised, aspiration of bacteria from the URT into the lungs can result in severe respiratory infection. 27,103 In addition to pathogens, the commensal microbiota can also be a reservoir of antibiotic resistance and virulence genes. 35,76 As many commensals and pathogens of the respiratory tract are naturally competent, namely, have the ability to take up exogenous DNA (eg, Streptococcus, Haemophilus, Neisseria), 56,58 under selective pressure, antibiotic resistance can likely spread rapidly through this community.
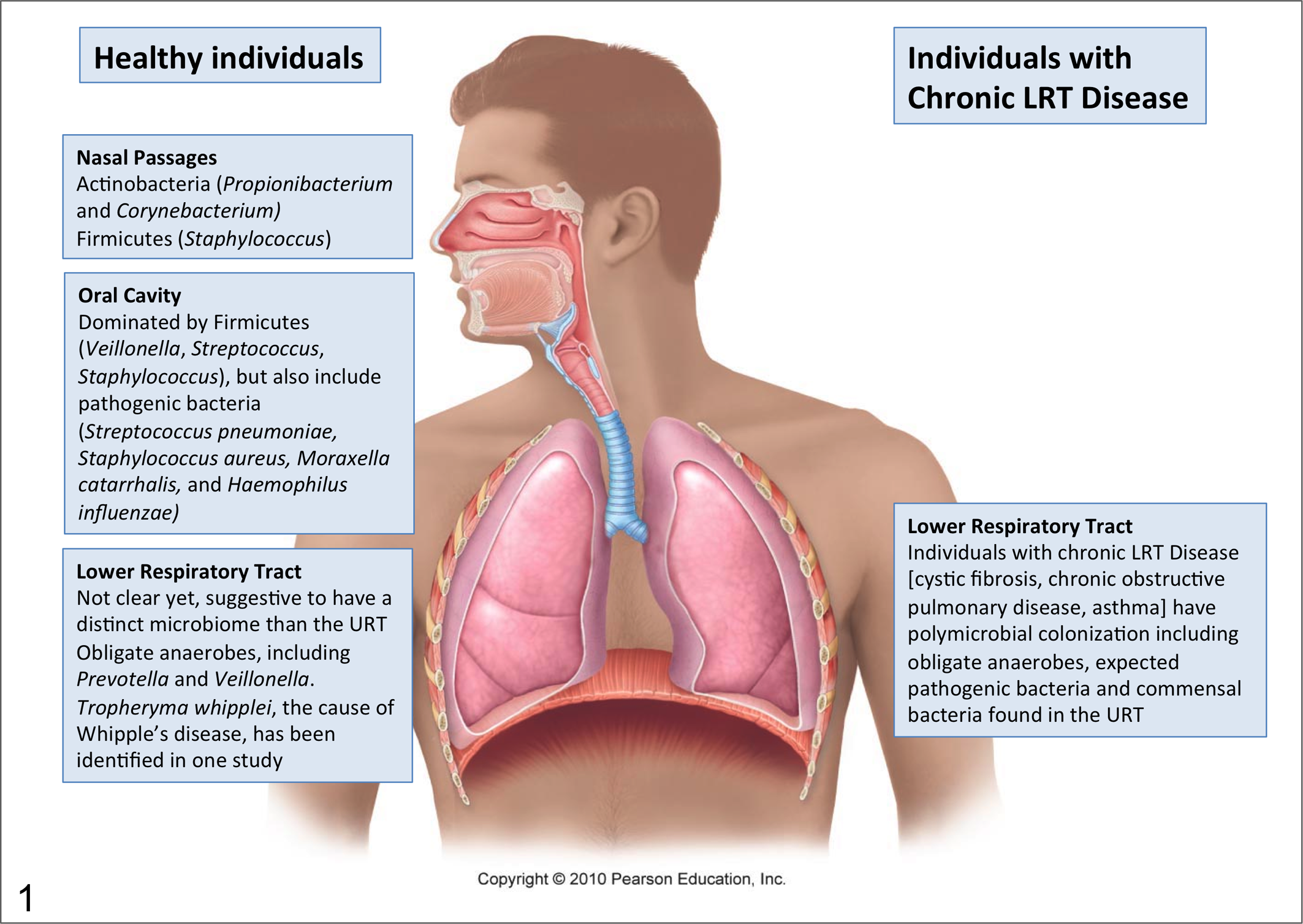
Different microbial communities colonize the upper and lower respiratory tract in humans. Individuals with chronic infections are colonized by obligate anaerobes, pathogenic bacteria, and bacteria found in the upper respiratory tract. Modified from Marieb EN, Hoehn K. Human Anatomy and Physiology. 8th ed. 2010. Printed and electronically reproduced by permission of Pearson Education, Inc, Upper Saddle River, New Jersey.
Most studies on the URT microbiota have focused on healthy adults. 14,24,74 However, it is important to consider that the groups most susceptible to respiratory infections are the very young and the elderly. 3,60 Bogaert et al 10 looked in depth at children under 2 years of age in the Netherlands and observed 4 clusters, 3 dominated by a single operational taxonomic unit (Moraxella, Haemophilus, or Streptococcus) and 1 that was mixed. 10 Similar results were observed in a recent study of healthy children from a Canadian city, except that a Haemophilus-dominant group was not observed. 110 The microbiota of young children (<2 years) was distinct from that of adults, while that of older children in this study (up to 4 years) was similar to adults, although too few individuals were sampled in this age group to make strong conclusions. The nasopharynx of adults was colonized with larger proportions of Firmicutes and Actinobacteria, whereas younger children had similar groups, but less diversity of their microbiome, and larger proportions of Proteobacteria, Firmicutes, and Enterococcus. These results suggest changes in composition of the URT microbiome that may parallel maturation of the immune system as is observed in the digestive tract. 66,95 Quantitative culture also suggests that the total bacterial burden is much higher in young children and decreases after the first few years of life. 110 Interestingly, the oropharyngeal microbiota in this study was more similar between children than adults. Seasonal variations in microbiota in young children may also contribute to susceptibility to pediatric tonsillitis and ear infections. 10
A complementary study of the microbiota of the URT of the elderly (age >65) showed marked differences between the microbiota of this age group compared to younger healthy adults (age 18–43). 125 While there is a clear distinction between the nasal and oropharyngeal microbiota in younger adults 14,51,74 and even young children, 111 this topographic difference is lost in the elderly. 125 Overall, there was an increased relative abundance of streptococci (specifically species of the Streptococcus salivarius group but not Streptococcus pneumoniae) in the oropharynx of elderly.
These studies on the URT microbiota of the young and elderly, the group most vulnerable to respiratory infections, suggest that the changing microbiota, in combination with a maturing and senescing immune system in the very young and elderly, respectively, play a role in this susceptibility. This will not be unique to humans but likely occurs in all mammals.
Microbiome studies in companion animals focusing on the upper and lower respiratory tract in young, adult, and aged animals are currently lacking. Of the few studies published to date, Moraxella sp. was frequently identified in samples from the nostril and oral cavity of healthy dogs, 99,112 accounting for approximately 33% of the bacteria colonizing the nostril. Bacteria in the genus Moraxella were also isolated from oral swabs from healthy dogs 61 and from bronchial samples from dogs with tracheal collapse. 57
The Lower Respiratory Tract Microbiome
The lower airways have been considered to be a sterile site in healthy individuals or more appropriately an effectively sterile site. During normal breathing, the lower airways in an adult human are exposed to 105 microorganisms per day through aerosols. These were thought to be effectively dealt with by the airway immune defense. 9,21 However, this continued exposure to microbes has prompted investigations of the lower airways in healthy individuals and led to questions of whether there may be a viable microbial colonization of the lungs. The challenge for studies addressing these questions is contamination of the sample during bronchoscopy and the suggestion that the diverse community observed represents contamination rather than true colonization of the lower airways. To address this, oral microbiota profiles were compared to bronchoalveolar lavage (BAL). 6 If there were a true lower airway community, it is predicted that the composition would look different than the oral microbiota in the same individuals. Even if the lower airways were colonized from the upper airways, as a different ecological environment, some but not all species would be similar, and they would differ in their relative abundances. The data from recent human studies suggest a lower airway microbiome, but it was certainly not convincing in all samples. 77,90 These 2 studies compared oral washes to BAL fluid and observed distinct differences in the microbiome composition, with Enterobacteriaceae, Haemophilus, Methyllobacterium, and Ralstonia being disproportionally more abundant in the lungs. Surprisingly, there were no statistical differences in the microbiota between healthy individuals and smokers. Another unexpected finding was the prevalence of Tropheryma whipplei in the samples from the lower airways. 77,90 T. whipplei is the causative agent of Whipple’s disease, a rare systemic disease primarily associated with gastointestinal infections. 28 The clinical significance of these findings is unknown. Another study examining the LRT microbiota using similar comparisons but carried out on BAL obtained through the nasal route to minimize oropharyngeal contamination observed URT microbiota in some but not all individuals. 104 In other samples, bacterial DNA was detected but most likely represented low-level DNA contaminants in the BAL fluid. More importantly, low-level inflammation was detected in the BAL fluids when the URT microbiota signature was recovered, providing further support for the presence of bacteria in the lower airways. No T. whipplei was reported in this study.
These studies provide evidence for presence of bacteria in the LRT of healthy human individuals. They do not provide evidence of a stable unique lower airway microbiome, however. Indeed, the results of Segal et al 104 are consistent with these microbiota in the LRT being transient in healthy individuals. Furthermore, like all molecular studies, they provide evidence for bacterial DNA and not necessarily viable organisms. Bacterial products would be sufficient to induce an immune response independent of the presence of viable organisms. However, these studies do indicate that the LRT are constantly exposed and any compromise to host defense or immune suppression could result in rapid development of LRT infections. Stress-mediated suppression of the immune system in shipping fever of cattle is an example. 46 LRT infections are often responsible for high morbidity and mortality in both the developed and developing world. For patients with severe pneumonia, even when carefully diagnosed, an etiological agent (bacterial or viral) is identified in less than 50% of cases. 6,12,84 Commensal microbiota are often recovered but dismissed clinically as contamination. It is possible that we may be underestimating the pathogenic potential of organisms we dismiss as commensals of the upper repiratory tract.
In the case of chronic lower airway disease (eg, in cystic fibrosis [CF], 80 chronic obstructive pulmonary disease, and asthma) and even in more acute infections in humans, it is now widely accepted that the lower airway colonization is polymicrobial (Fig. 1). 9,47,79,97,120 These communities include many members of the upper airway microbiome in addition to the expected LRT pathogens. In disease states, such as in CF, several studies have shown that the sputum or BAL microbiome is distinct in microbial composition and relative abundance compared to the oral microbiome, 100 ruling out contamination as a major source of these organisms. Importantly, obligate anaerobes make up a significant portion of these lower airway microbiomes. Even in the studies of the LRT microbiome from healthy individuals, obligate anaerobes (eg, Prevotella and Veillonella) are among the most common bacteria detected. 77,90,104 The role of these additional microbes in disease is not understood. Using animal models, it has been shown that bacteria isolated from the airways of CF patients, which have little or no pathogenic potential, can synergize with pathogens to enhance virulence, suggesting a role for polymicrobial interactions in disease progression. 22,105 This synergy involves microbe-microbe interactions that result in the modulation of bacterial virulence factor gene expression. 105 It is likely that most lower airway infections are polymicrobial. 80,107 Whether this will influence the progression of disease is not known.
The Microbiome in the Digestive Tract
The Digestive Tract Microbiome in Health
An estimated 1010 to 1014 microbial cells are present in the intestine of mammals, which is approximately 10 times more than the number of host cells. Sequence analysis of 16 S rRNA genes has revealed a highly complex ecosystem within the human, 123 canine, 43,44,114 and feline 44 GI tract. This complex intestinal microbiota has a significant impact on health and disease. Several reviews have covered the human digestive tract microbiome in depth. 64,109,123 This section will focus primarily on the digestive tract microbiome of companion animals.
The exact number of bacterial species in the digestive tract remains unknown, mostly due to the technical difficulties in accurately describing this complex ecosystem. Generally, the composition of the gut microbiota is to some extent similar across humans, dogs, and cats. At least 200 bacterial phylotypes are estimated to be present in the canine small intestine, while the canine colon harbors a few hundred to thousand bacterial phylotypes. 44,114 The Firmicutes and Bacteroidetes are the major bacterial phyla in the gut of dogs and cats (Fig. 2). 2,3 Less abundant phyla in dogs are Proteobacteria, Actinobacteria, Spirochaetes, Fusobacteria, Tenericutes, Verrucomicrobia, Cyanobacteria, and Chloroflexi. The phylum Firmicutes comprises many distinct bacterial groups. Of those, Clostridium clusters XIVa and IV are the most abundant and encompass many important short-chain fatty acid–producing bacterial groups (ie, Lachnospiraceae, Ruminococcus, Faecalibacterium, Dorea). There is a gradual increase in species richness and abundance of bacteria from the small to the large intestine. The canine stomach harbors a microbiome that is often dominated by Helicobacter spp., which comprised 99% of 16 S rRNA sequences in one study. 34 Bacterial counts in the canine and feline duodenum typically range from 102 to 105 colony forming unit per gram (cfu/g) of content. However, up to 109 cfu/g have been reported in healthy dogs and cats. 37,59 The total bacterial count in the colon ranges between approximately 109 and 1011 cfu/g.
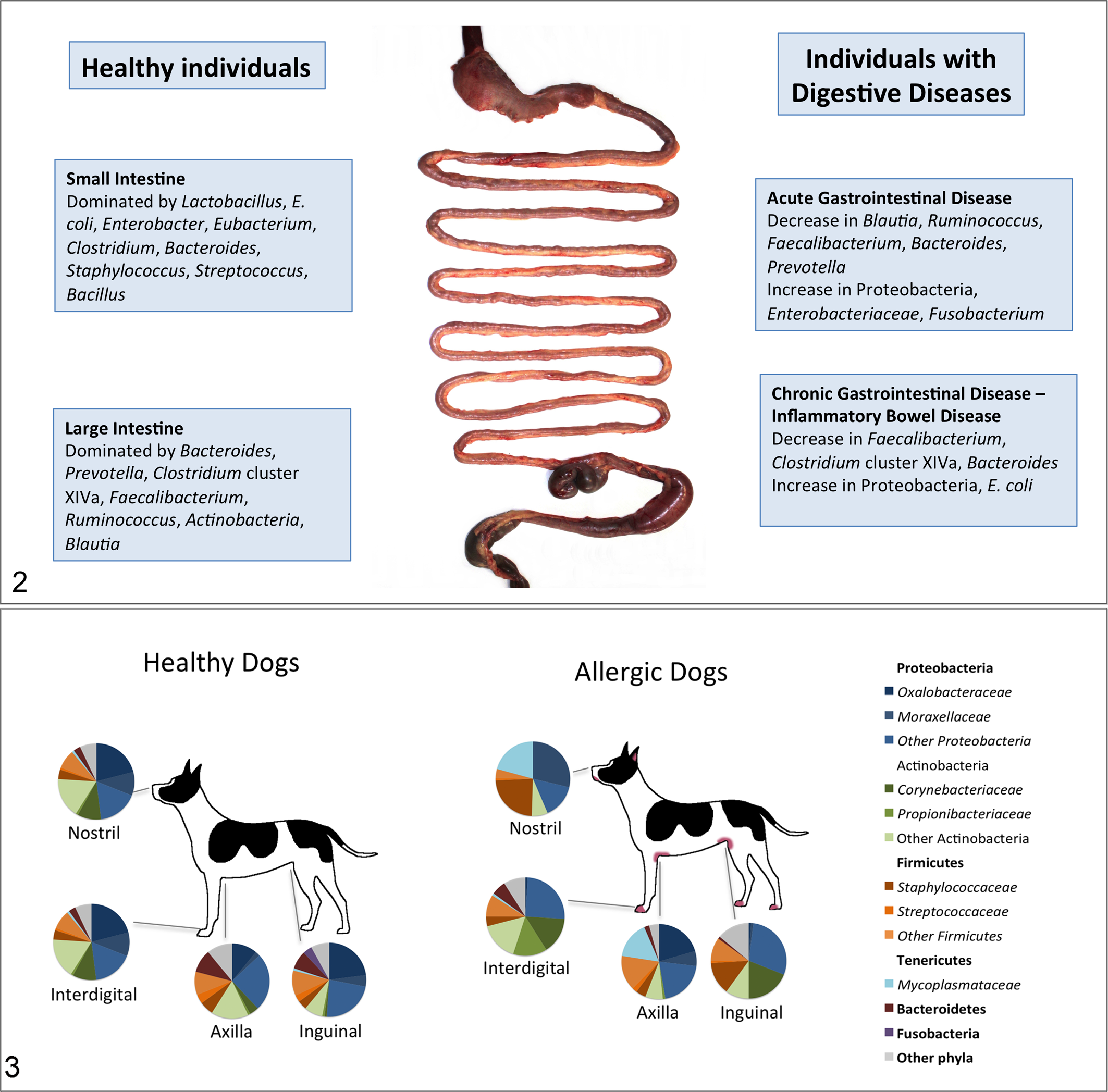
The most common bacteria identified in the digestive tract and feces in healthy dogs and cats and those with acute and chronic gastrointestinal disease.
The digestive tract harbors not only bacteria but also various fungi, archaea, protozoa, and viruses. Recent studies have provided more in-depth analysis about the diversity of these microorganisms in healthy individuals, but their interactions, influences on the host, and role in disease remain unclear. Analysis by fluorescence in-situ hybridization (FISH) and DNA shotgun sequencing analysis of fecal DNA obtained from healthy humans and canines have estimated that bacteria make up approximately 98% of all sequences, and fungal organisms and archaea together make up <2%. 102,121 Similarly, DNA shotgun sequencing of feline fecal samples revealed 97% of sequences as of bacterial origin, 1% from eukaryota, 0.1% from archaea, and 0.1% from DNA viruses. 122
Pyrosequencing of the fungal 18 S rRNA gene reported Aspergillus and Saccharomyces as the most abundant fungal genera in pooled fecal samples of cats. 44 In another study, 76% of dogs were positive for fungal DNA on duodenal biopsies. 118 In this study, 51 different fungal phylotypes were described, with Pichia, Cryptococcus, Candida, and Trichosporon being most frequently observed. A complex fungal microbiome has been also described in fecal samples of dogs. 32 Five distinct fungal phyla were identified across healthy dogs and dogs with diarrhea. The phyla Ascomycota (97% of fungal sequences) and Basidiomycota (1%) predominated. Dogs harbored a median of 28 (range, 4–69) fungal genera, with Candida as the most commonly observed genus in this study.
The intestinal microbiome exerts benefits on the host through many mechanisms. Commensal bacteria provide a defensive barrier against transient pathogens. They aid in nutrient digestion and provide important metabolites for the host. Furthermore, microbes have an important function in modulating the immune system of the host. 113 Studies using shotgun DNA sequencing have described functional properties of the intestinal microbiota by providing information about the microbial genes present in the intestine. For example, the most abundant functional gene categories of the microbiome in cats pertained to microbial carbohydrate and protein metabolism (13%–15% and 6%–8% of the feline metagenome, respectively), DNA metabolism (7%–8%), virulence factors (6%–7%), amino acid metabolism (6%–8%), cell wall and capsule (7%), and cofactors, vitamins, prosthetic groups, and pigments (6%). 8,122
The Microbiome in Gastrointestinal Diseases
Several studies have reported altered microbial communities in acute and chronic GI diseases of dogs and cats. 5,17,20,38,55,88,89,115,117,119,126 These microbial changes are accompanied by underlying susceptibilities in the innate immune system of dogs and cats with chronic enteropathies, such as idiopathic IBD, further demonstrating the relationship between gut microbiota and host health. 55,62,63,78,86
It is been shown that the canine microbiota changes with diarrhea, with the most profound changes in dogs with acute hemorrhagic diarrhea (AHD), 117 and characterized by significant decreases in Blautia, Ruminococcaceae including Faecalibacterium, and Turicibacter spp., and significant increases in genus Sutterella and Clostridium perfringens compared to healthy dogs (Fig. 2). Analysis of selected bacterial groups by quantitative PCR in fecal samples from healthy dogs, dogs with chronic enteropathies (CE), and dogs with AHD showed pronounced decreases in Faecalibacterium, Turicibacter, and Ruminococcaceae, with significant increases in E. coli and C. perfringens in CE and AHD. 83 Particularly, Faecalibacterium spp. appears frequently depleted in canine GI disease. 83,88,101 This bacterial group also correlated with improvement in the clinical IBD activity index, suggesting that it might be a useful marker for monitoring improvement of fecal dysbiosis. 101,117
In canine and feline IBD, patients are often presented with persistent or intermittent GI signs. A diagnosis of IBD is reached by excluding other chronic enteropathies, such as those that respond to treatment with dietary change, antibiotics, or glucocorticoids. There is now strong evidence that besides environmental factors and genetic alterations, the gut microbiota plays an important part in the pathogenesis of IBD, mostly due to imbalances in the GI microbial populations, namely, microbial dysbiosis. A decrease in the phyla Firmicutes and Bacteroidetes and an increase in Proteobacteria and Actinobacteria have been associated with IBD. 94 In dogs with IBD, the mucosa-adherent microbiota of the small intestine is primarily comprised of Proteobacteria, especially E. coli-like organisms 126 or Pseudomonas, 119 with lower proportions of Firmicutes and Bacteroidetes compared to healthy dogs. 115
Boxer dogs and French Bulldogs with granulomatous colitis also have dysbiosis of their intestinal microbiome. Microbiota analysis based on sequencing of 16 S rRNA genes in combination with FISH has revealed invasive E. coli bacteria present in multifocal clusters within macrophages in the colonic mucosa. 106 These dogs often respond well to antibiotic treatment, with remission of clinical signs, suggesting a causal relationship between these bacteria and the disease. 106 Given the association of granulomatous colitis with invasive bacteria, FISH can be utilized as a diagnostic tool on intestinal biopsies to better guide treatment decisions. 81
To date, limited information is available about microbial dysbiosis in feline IBD. 98 In 1 study based on FISH analysis, Enterobacteriaceae were increased in duodenal biopsies of cats with IBD, with a significant relationship between severity of histological inflammation and increased numbers of bacteria. 55 In another study evaluating number of bacteria using FISH, fecal samples from cats with IBD had lower numbers of Bacteroides and Bifidobacterium and higher numbers of Desulfovibrio compared to healthy cats, 53 although another similar study did not identify differences between healthy cats and those with GI disease. 2 A recent study, using next-generation sequencing of 16 S rRNA genes, described the fecal microbiome of cats with acute or chronic diarrhea. 116 As in other animal species, the major changes were in increases in Enterobacteriaceae, with concurrent decreases in Bacteroidetes and members of Clostridium Cluster XIVa. Furthermore, these microbial changes were accompanied by altered bacterial functional gene content: altered metabolism of glycosphingolipids, fatty acids, biotin, tryptophan, ascorbate, and aldarate.
The prevalence and identification of fungal organisms have also been evaluated in dogs with chronic enteropathies. 118 Besides the fungal taxa observed in the digestive tract of healthy dogs, those with gastrointestinal disease also harbored opportunistic fungal pathogens. 118 Another study that evaluated the fungal microbiome in healthy dogs and those with gastrointestinal disease did not identify significant differences in the relative proportions of fungal communities between healthy and diseased dogs. 32 Therefore, additional studies are needed to elucidate the importance of fungi on intestinal health and disease of animals.
It is likely that dysbiosis of the microbiome alters the metabolites produced by gut bacteria, which in turn may dysregulate innate and adaptive immune responses. This may ultimately lead to inflammation and/or loss of protection against infection. Certain gut bacteria can produce toxins, such as ammonia, D-lactate, endotoxin (lipopolysaccharide), and exotoxin (enterotoxin), which can further aggravate intestinal lesions. On the other hand, gut bacteria also have an important protective role in gastrointestinal homeostasis. For example, Lachnospiraceae, Ruminococcaceae, and Faecalibacterium, important producers of short-chain fatty acids (SCFA), have been found to be depleted in GI disease. The reduced production of SCFA may impair the capability of the host to down-regulate intestinal immune responses. Initial studies performed in dogs with intestinal disease have demonstrated an association between intestinal dysbiosis and altered metabolism. In 1 study, analysis of serum metabolites in dogs with IBD suggested an association between fecal dysbiosis and altered energy metabolism and an increase in oxidative stress of affected dogs. 89 Another study evaluated serum and fecal metabolites in dogs with acute diarrhea and reported changes in tryptophan metabolism in diseased dogs. 42 Further studies are warranted that correlate microbial dysbiosis with functional changes within the intestine of companion animals.
The Skin Microbiome
The Skin Microbiome in Health
As seen in other organ systems, the skin is a complex ecosystem and is colonized by a wide variety of microorganisms, including bacteria, fungi, and viruses. 41 The normal skin microbiota is necessary for optimal skin fitness, modulating the innate immune response and preventing colonization of potentially pathogenic microorganisms. 124
Studies using sequencing of 16 S rRNA genes have revealed that the skin surfaces of humans and companion animals are inhabited by a highly diverse microbiota that was previously not appreciated by culture-based methods. 40,41,67,69 Furthermore, there are topographic differences in the various skin surfaces, with the microbiota from similar skin locations of different people being more closely related than different skin locations from the same individual. 41 Temperature, pH, moisture, environmental contact, and contact with mucous membranes are some of the factors that may influence the variability of bacterial abundance and distribution on the skin. 41,108 In humans, Propionibacterium predominantly colonize the sebaceous areas, Staphylococcus and Corynebacterium are commonly found in moist areas, and gram-negative organisms are more likely to colonize dry skin areas such as the forearm or leg. 41
The skin microbiota also changes with age, with infants having significantly different microbial populations than adults. For instance, the relative abundances of Staphylococcus and Streptococcus in the forehead skin decreased, while Propionibacterium inhabiting the forehead increased with age. 13 The composition of the skin microbiota in humans can also be altered by contact, for example in sports involving skin-to-skin contact. 87 Such studies, evaluating changes related to age and environment, are currently lacking in companion animals. The diversity of the skin microbiota in adult humans is also influenced by cohabitation with pets, in particular dogs, 108 with people who cohabit with dogs sharing a more similar microbiota than those who do not own dogs. In contrast, ownership of indoor cats did not appear to influence the diversity and composition of the skin microbiome among cohabiting adult individuals.
The skin microbiome also extends beyond the epidermal surface. By utilizing 16 S rRNA sequencing, Gram staining, and FISH analysis, bacterial DNA and antigen were detected within the dermis and subcutaneous tissues in humans. 91 However, due to the methods used in this study, it was not possible to conclude whether live bacteria inhabit these regions.
The human skin also harbors a diverse fungal microbiome. 29 The genus Malassezia was most abundant in all skin regions, with 11 of the 14 known Malassezia species being identified among skin sites. The plantar heel was the most diverse site with higher representation of different fungal genera, including Malassezia, Aspergillus, Cryptococcus, Rhodotorula, and Epicoccum.
A recent study evaluated the diversity of the skin microbiota in different cutaneous and mucocutaneous regions in healthy dogs. 99 Similar to humans, 41 the different skin sites from each dog were inhabited by a variable and unique microbiome, with significant individual variability between samples from different dogs and between different skin sites. 99 A large number of previously uncultured or rarely isolated microbes were identified, demonstrating that the skin of dogs is inhabited by diverse microbial communities. Higher microbial diversity was observed in the haired skin (axilla, groin, periocular, pinna, dorsal nose, interdigital, lumbar) compared to mucosal surfaces or mucocutaneous junctions (lips, nose, ear, and conjunctiva). 99 The nostril and conjunctiva showed the lowest, while the axilla and dorsal aspect of the nose showed highest microbial diversity. 99 On average, around 300 different bacterial genera were identified on the canine dorsal nose. The most abundant phyla across all surfaces were Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes (Fig. 3). The family Moraxellaceae was most abundant in the nostril. 99
The Skin Microbiome in Cutaneous Diseases
In many skin conditions, it remains unclear if changes in the microbiome play a causal role in skin diseases or are rather the result of the disease. 129 In humans with atopic dermatitis (AD) and psoriasis, the changes in the cutaneous microbiome have been proposed to be the result of an altered epidermal barrier function, Toll-like receptor 2 defects, decreases in antimicrobial peptides, and/or increased expression of extracellular matrix proteins. 18 These mechanisms are thought to be responsible for an increased abundance of Staphylococcus aureus and susceptibility to staphylococcal infections in AD patients. 18 Infection with S. aureus correlates with clinical severity in humans with AD. 45 Recurrent infections with Staphylococcus sp. are very common in AD dogs, and in some dogs bacterial products can also trigger lesions of AD, possibly due to an altered epidermal barrier function. 19,85 A marked reduction in microbial diversity and an increase in cutaneous S. aureus were observed in children during AD flares, and it was proposed that these changes precede an increase in the severity of AD. 68 Besides S. aureus, the skin commensal S. epidermidis was also increased during non-treated flares. The authors suggested that the commensal relationship between these 2 bacteria could enhance common resistance to antimicrobial peptides or enhance binding to exposed extracellular matrix proteins in inflamed skin. In these atopic children, antimicrobial or anti-inflammatory medications (hypochlorite baths) decreased the relative abundance but did not eliminate S. aureus. The changes in the microbial diversity during flares were reversed even before clinical improvement was seen. 68
Strain differences rather than presence or absence of certain species may explain the onset of certain diseases. It is well known that Propionibacterium acnes is associated with acne. 30 Remarkably, this bacterium is 1 of the main commensals identified in sebaceous areas, 41 and the relative abundances of P. acnes are very similar among healthy individuals and those with acne. 30 Individuals with acne are colonized with different strains of P. acnes carrying virulence genes previously identified in other organisms. These likely contribute to the virulence and pathogenicity of P. acnes in individuals with acne. 30 Human patients with psoriatic plaque have a different microbiome compared to healthy individuals, with increased Corynebacterium, Propionibacterium, Staphylococcus, and Streptococcus. 4
Studies utilizing bacterial culture have shown that the nasal mucous membranes of atopic humans 10 and dogs 12 are more often colonized with S. aureus and S. pseudintermedius, respectively, compared to healthy individuals. In contrast, genomic analysis of the microbial diversity in the nose of humans with AD did not reveal any association between the relative abundance of S. aureus and disease severity, as the relative proportion of S. aureus did not change prior to, during, or after flares. 9 On the other hand, the antecubital (in front of the elbow) and popliteal creases, common sites for skin lesions of AD in children, were markedly dominated by S. aureus during flares. 9 It was proposed that the site specificity of certain skin bacterial communities are important in the initiation and perpetuation of certain skin diseases. 9
In most dogs with AD, primary skin lesions are characterized by intensely pruritic erythematous macules and patches, mainly affecting the front and hind paws, axilla, and inguinal region. 25 These lesions usually reflect the chronic course of the disease. Concomitant bacterial and/or fungal infections, most commonly by S. pseudintermedius and Malassezia sp., 26 often result in exacerbation of skin lesions and development of papules, pustules, and crusts. 93 The skin microbiome colonizing the haired skin of dogs with allergic skin disease was evaluated and showed decreased bacterial diversity compared to the same skin sites (axilla, groin, and interdigital skin) of healthy dogs. 99 Significant differences in bacterial taxa were also observed between allergic and healthy dogs, especially higher abundance of Betaproteobacteria in the skin of healthy dogs. The most frequently identified bacterial genera in the skin of allergic dogs included Alicyclobacillus, Bacillus, Corynebacterium, Staphylococcus, and Sphingomonas (Fig. 3).
The importance of the skin microbiome goes beyond its commensal or pathogenic capabilities. The skin microbiome also modulates the innate and adaptive immune responses. In a recent study conducted in mice to evaluate the relationship of the microbiome with complement signaling, it was demonstrated that blocking/inhibiting C5a receptor resulted in decreased microbial richness and diversity. 92 The skin microbiome can directly influence T-cell function and the local inflammatory response, besides promoting protective immunity against other pathogens, such as protozoa. 36
Conclusion and Future Directions
The studies presented here demonstrate that the interactions between host and microbes play an important role in health, susceptibility to infection, and the response to treatment of individual patients who are predisposed to respiratory, gastrointestinal, or skin diseases. We are only beginning to appreciate these complex relationships with the host and how they are influenced by various environmental factors, age, and disease.
Studies using culture-independent methods have revealed this complexity but have largely focused on the bacterial populations. Much less is known about viral and fungal contributions to health and disease states. There is a lack of comprehensive studies both in the human and veterinary literature evaluating the functional consequences of these alterations and how changes in the microbiota correlate with morphologic and metabolic changes in tissues. Examining these host and microbial interactions in the different organ systems in humans and animals will allow us to better understand the pathophysiology of diseases and how the microbiome contributes to initiation and/or aggravation of disease. Understanding the pathways by which the microbiota shape and/or are shaped by the immune system will allow the development of therapies, aimed in modulating microbial communities and their produced metabolites, and thereby reducing susceptibility to infections.
Footnotes
Author Contributions
All authors contributed equally for this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.