
Abstract
Rheumatoid arthritis (RA) is a chronic debilitating autoimmune disorder characterized by synovitis that leads to cartilage and bone erosion by invading fibrovascular tissue. Mouse models of RA recapitulate many features of the human disease. Despite the availability of medicines that are highly effective in many patient populations, autoimmune diseases (including RA) remain an area of active biomedical research, and consequently mouse models of RA are still extensively used for mechanistic studies and validation of therapeutic targets. This review aims to integrate morphologic features with model biology and cover the key characteristics of the most commonly used induced and spontaneous mouse models of RA. Induced models emphasized in this review include collagen-induced arthritis and antibody-induced arthritis. Collagen-induced arthritis is an example of an active immunization strategy, whereas antibody- induced arthritis models, such as collagen antibody–induced arthritis and K/BxN antibody transfer arthritis, represent examples of passive immunization strategies. The coverage of spontaneous models in this review is focused on the TNFΔ ARE mouse, in which arthritis results from overexpression of TNF-α, a master proinflammatory cytokine that drives disease in many patients.
Keywords
Lesions in rheumatoid arthritis (RA), a debilitating systemic autoimmune disease, are characterized as chronic destructive synovitis with predilection for small diarthrodial joints, especially those of the hands and feet. The pathogenesis of RA is complex and involves genetic predispositions as well as environmental factors. 21 Central to the pathogenesis is the activation of macrophages by autoreactive T cells, resulting in the release of key proinflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interleukins 1, 6, and 17 (IL-1, IL-6, and IL-17). 13 Immunotherapies targeting these master cytokines, their receptors, or their downstream signaling components have shown dramatic success and greatly improved disease management in patient populations that have access to these therapies. 13 Yet, the events that trigger the generation and recruitment of autoreactive lymphocytes in the first place are still emerging. For example, the role of human leukocyte antigen variants in promoting reactivity to citrullinated antigens has only recently been recognized as important in the initial pathogenesis. 6,12,46 Antibodies to citrullinated antigens are detectable in a variety of autoimmune diseases, including RA, indicating that citrullination plays a role in the process of epitope spreading 52 and the emergence of autoepitopes. 35,60,64 Likewise, citrullinated antigens are relevant in animal models of autoimmunity. 9,24,58 Neutrophils represent another example of hitherto underappreciated contributors to the pathogenesis of autoimmune diseases. The role of neutrophils in initiating and promoting autoimmunity to citrullinated nuclear antigens through the extrusion of neutrophil extracellular traps is being reevaluated. 70
Mouse models that recapitulate aspects of the pathogenesis in humans are an important tool for investigating such mechanisms in vivo and have been in use for many years. The purpose of this brief review is to summarize the key features of some of the more widely used induced and spontaneous models of immune-mediated joint disease (Fig. 1). For the appropriate selection of models according to features of pathogenesis in human RA, we recommend consulting the review by Vincent et al on behalf of the Arthritis Research UK Animal Models Working Group. 63 That article also provides useful information on features of osteoarthritis and briefly touches on important considerations for animal welfare and statistical powering of groups, areas that are not covered here. General practical considerations in the use of various rodent joint disease models are reviewed by Bolon et al. 3 Finally, for a general scoring scheme of histologic lesions, we refer to Caplazi and Diehl. 4 Besides this general scoring scheme, other, more narrowly defined morphologic scoring systems may be suited in situations where therapeutic agents selectively influence some lesion features. For the assessment of bone lesions, sophisticated quantitative methods based on histomorphometry are possible 47 but may require more advanced processing techniques, such as embedding of undemineralized tissue in methylmethacrylate. While histology remains the most widely used method for assessment of lesions in mouse models of RA, sophisticated in vivo or ex vivo imaging modalities exist that can complement or altogether replace histologic evaluation. 2,29,32,62,71
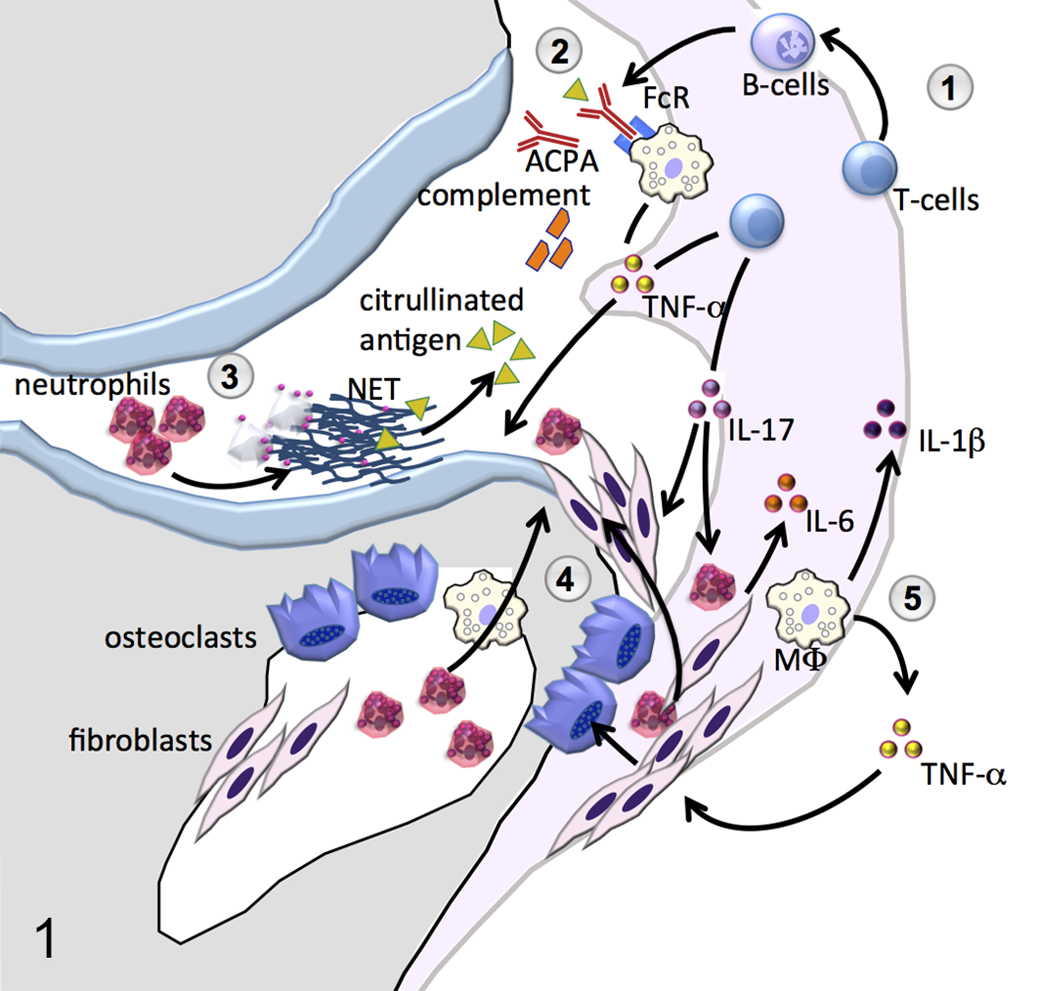
Key concepts shared between human rheumatoid arthritis (RA) and mouse models of RA. ① Autoreactive T cells providing help to B cells or releasing a variety of proinflammatory cytokines are an important component in the initiation and maintenance of lesions in RA as well as mouse models. 9,48 An intact adaptive immune system is not necessary for some models, however. Notably, T cells are not required for the induction of collagen antibody–induced arthritis. 37 The antigen-experienced T-effector/memory cell phenotype is prominent among T cells in the RA synovium. 8 ② Antibodies to citrullinated antigens (anticitrullinated peptide antibodies [ACPAs]) that are implicated in the pathogenesis of RA have been shown to be important in mouse models, including collagen-induced arthritis. 24,28 More broadly, antibody- and complement-mediated effects are important drivers in RA as well as mouse models. 33 ③ Neutrophils are abundant in murine autoimmune arthritis and contribute to the pathogenesis through the release of cytotoxic products and immunoregulatory mediators. In addition, neutrophils may promote autoimmunity by formation of neutrophil extracellular chromatin traps (NETs) and the associated promotion of ACPA. 11,70 ④ Cartilage and bone injury is driven by the formation of an inflammatory pannus that classically invades the joint from the capsular angle. In mouse models, however, substantial amounts of pannus also extend from the bone marrow cavity subjacent to the articular cartilage (Figs. 3, 9, 13). ⑤ Macrophages (MΦ) infiltrating the synovium are central in the pathogenesis of RA, serving as an important source of key proinflammatory cytokines such as interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α). Furthermore, macrophage-derived cytokines and their effects on synovial fibroblasts are essential for the differentiation of osteoclasts and osteolysis. Bones are shown in gray, articular cartilage in light blue, and synovium and adjacent soft tissues in light pink.
Systemically Induced Mouse Models of RA
Based on the method of induction, systemically induced models can be divided into 3 groups: those elicited by active immunization, those elicited by passive immunization, and those elicited by administration of irritant chemicals resulting in chronic inflammation.
Collagen-induced arthritis (CIA) is the archetypical model of RA induced by active immunization. First developed in rats, 59 the model was later adapted to the mouse 16 and is now commonly used in both species, with the choice of rodent dependent on the scientific questions and types of therapies to be tested. Rats or mice develop an acute to subacute monophasic erosive polyarthritis after immunization with collagen II (but not other collagens) from many species. Type II collagen is the major constituent collagen form of articular cartilage, and immunoreactivity to type II collagen can be identified in some RA patients. 7,56 Many important features of human RA are recapitulated in mouse CIA, including the presence of rheumatoid factor or anticitrullinated peptide antibody. 51 At least in the mouse, a Th17-driven response is essential for the production of lesions. 41,50 The effects of interferon γ are divergent 51 with inhibitory as well as stimulatory effects on development of disease. Overall, however, interferon γ attenuates CIA. 22,51
Practical aspects of working with the mouse CIA model have been reviewed elsewhere 66 and so are only briefly summarized here. A typical induction protocol might use heterologous (eg, bovine) collagen II for immunization of mice from a strain with permissive major histocompatibility complex (MHC haplotype q, H-2q; eg, DBA/1). For induction, a subcutaneous injection of type II collagen suspended in complete Freund adjuvant on day 0 is followed by a booster of type II collagen in incomplete Freund adjuvant on day 21. Effects of experimental treatment can then be tested by applying a prophylactic design (treatment initiated before the onset of clinical signs at around day 20 to 30) or a therapeutic design (treatment initiated after the onset of clinical signs on animals selected for presence of clinical signs and then randomized into treatment groups).
Effects can be studied longitudinally in vivo using noninvasive assessments, such as clinical arthritis score, paw volume, caliper measurements of ankle thickness, or imaging modalities (eg, micro–computed tomography). Parameters determined in vivo can be complemented by terminal assessments, which include imaging (micro–computed tomography, benchtop radiography, magnetic resonance imaging), histology, or biochemical assays to determine tissue cytokine concentrations or gene expression levels. Paws selected for histology are fixed in formalin, decalcified, and processed to sagittal hemisections representing larger (eg, tarsal, carpal) as well as smaller digital joints. 3 Some investigators prefer to embed the hind paws in the sagittal plane, whereas forepaws are oriented horizontally. In addition to the usual considerations for histology, particular attention should be given to sample preparation. Important steps include even and precise sagittal trimming, complete dehydration during processing before paraffin infiltration, flat and level embedding of hemisections in paraffin, and slow sectioning of the embedded hemisections to prevent loss of bones or dislodgement of joints from the block. During the entire process of sectioning, blocks need to remain adequately hydrated, notably during the facing of blocks to a plane where larger and smaller joints are adequately represented. Dry facing of tissue blocks will result in loss of large segments of bone or entire joints from the blocks.
Experience with mouse CIA has shown that there is considerable individual variability with respect to incidence, synchronicity, and distribution of lesions. For example, it is not unusual (in up to 30% of animals in some of our studies) to find marked differences in lesion severity between paws (eg, spared paws, unilateral lesions). It is therefore beneficial to examine all limbs in each individual. Conversely, if limbs from a single animal are parsed out for different end points (eg, cytokine analysis, isolation of cells) in addition to histology, caution should be exercised in interpreting discrepancies of findings at an individual animal level. These caveats notwithstanding, lesions in mouse CIA are usually progressive and quite severe with extensive cartilage injury and bone remodeling 69 (Figs. 2–7). In the course of CIA, as in a number of other RA models, arthritogenic antibodies are produced and can be used for disease induction by passive immunization. 38
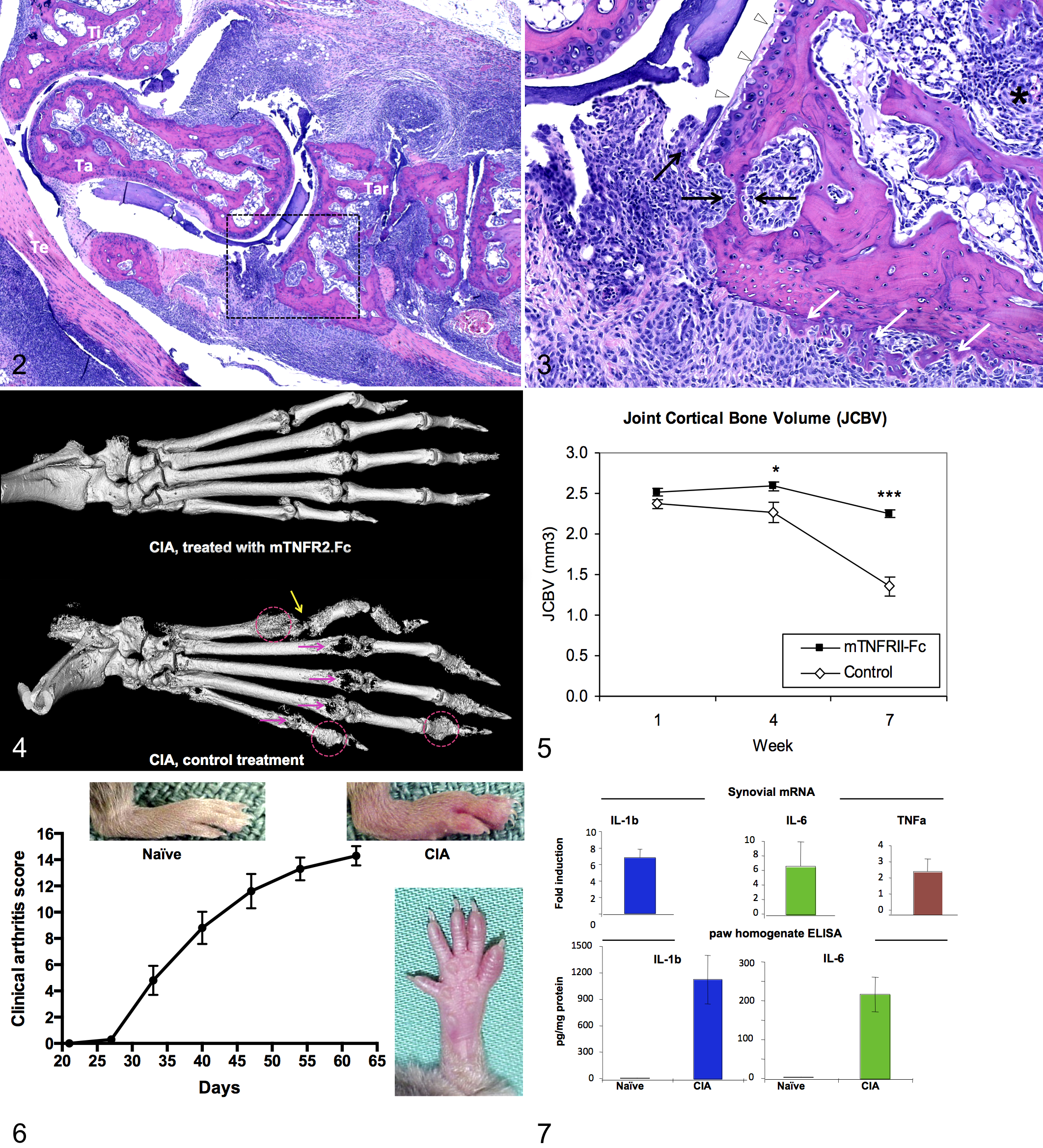
Collagen-induced arthritis (CIA), day 63 postinduction, tarsal region, sagittal section, mouse (DBA/1J). Hematoxylin and eosin.
A widely used example of arthritis induced by passive immunization is the direct derivative of CIA, collagen antibody–induced arthritis (CAIA). 39,40 In this model, arthritis is induced by systemic administration of mixtures of antibodies that target various epitopes of type II collagen. Clinical and histologic characteristics of CAIA (Figs. 8 and 9) are similar to those seen in CIA 17,23 and include important features of human RA, such as inflammatory synovitis, formation of pannus (an aggressive fibrovascular tissue that invades the joint), cartilage degradation, and bone remodeling, although acute components (eg, infiltration with neutrophils) prevail when compared to CIA. One particular benefit of CAIA over CIA is that the model is amenable for use in strains or genotypes not suitable for CIA. CAIA can be induced by serum transfer from arthritic mice 53 or human RA patients. 68 It is more practical, however, to use commercially available cocktails of monoclonal antibodies targeted to various regions of type II collagen. 40,57 Efficiency of disease induction using commercial or in-house generated reagents varies with reagent lots, mouse strain used, and environmental variables. CAIA, in contrast to CIA, is a fast model (peak disease within 8 days) and has a high degree of synchronicity in disease onset. After peak disease, clinical arthritis typically subsides within about 1 month 39 with concordant regression of histologic lesions. CAIA requires immune complex formation and complement activation, 1 but induction of arthritis is B cell and T cell independent and does therefore not recapitulate the complexity of immune and tissue remodeling responses during human RA. 37
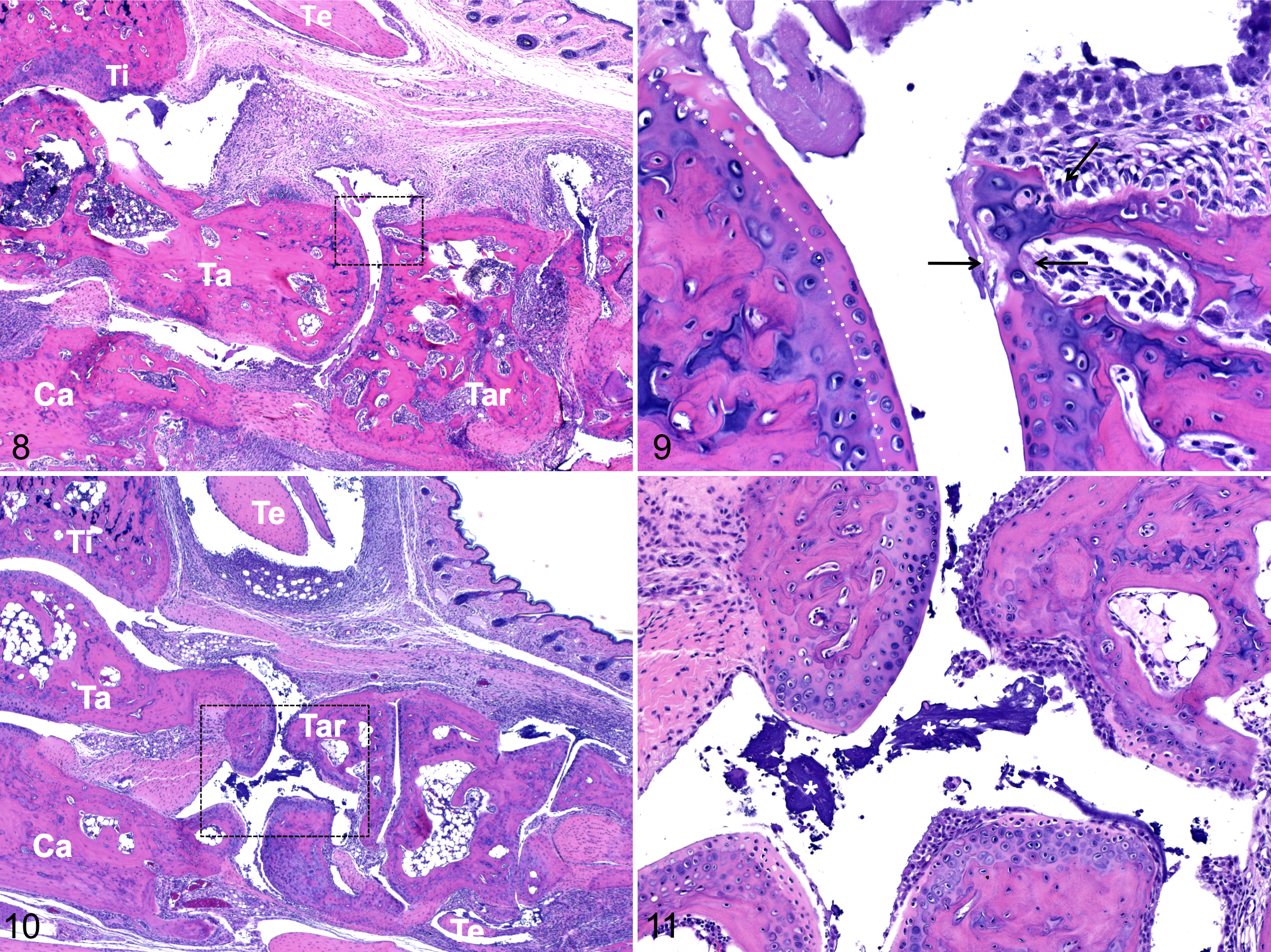
Conceptually similar to CAIA, K/BxN antibody transfer arthritis is another model that makes use of induction with arthritogenic antibodies; however, in this model, pathogenic antibodies are derived from K/BxN mice, which spontaneously develop severe arthritis. 34 K/BxN mice have a transgenic T-cell receptor that recognizes a peptide of G6PI (glucose-6-phophoisomerase) as an autoantigen in the context of MHC-II (I-Ag7). In this Th2-driven model, sustained T-cell help to B cells leads to the production of high-affinity IgG1 antibodies against G6PI in large quantities. 34 These antibodies are arthritogenic even after transfer to nontransgenic C57/Bl6 recipients, 27,31,34 although disease kinetics and characteristics vary by recipient strain. 34 K/BxN antibody-transfer arthritis has a rapid onset, occurring within 2 days after induction. Depending on the amount of antibody transferred, clinical signs and histologic lesions (Figs. 10 and 11) peak within 7 to 14 days and wane by day 21, although residual lesions (eg, ankylosis) may persist. Anti-GPI antibodies, neutrophils, macrophages, TNF-α, IL-1, and complement contribute to the pathogenesis in K/BxN arthritis. 38,48 The importance of neutrophils for the early pathogenesis of RA is highlighted in this acute model by the requirement of intact neutrophilic response for the development of disease. 67
Both of the antibody-transfer models described above are useful because they are applicable also to genetically engineered mice, in which arthritis might otherwise be difficult to induce. Thus, it becomes possible to study the influence of specific genes of interest in the context of a relevant RA model as exemplified in Sun et al. 54 The ability to dissect early disease mechanisms, coupled with the preponderance of acute lesions in affected joints, makes these passive immunization models favorite platforms for investigating potential new therapeutic targets.
Polyarthritis in the Context of Chronic Inflammation
The principle that polyarthritis can emerge on the background of chronic inflammation is applied by use of oily adjuvants in many of the RA models, including CIA as outlined above. The classical model is adjuvant-induced arthritis by intradermal injection of complete Freund adjuvant. Rats are predominantly used in this model, 65 but mouse variants exist also. 14,42 Pristane, a saturated alkane, is an example of a substance that by itself may induce arthritis in the context of chronic systemic inflammation. Arthritis was originally noted as a concurrent lesion in a murine model of plasmacytoma induced by intraperitoneal injection of pristane. 44 Based on this observation, pristane-induced arthritis was developed as a model. Because the response to TNF-α targeted therapy is limited in pristane-induced arthritis, this model is valuable for the study of therapies relevant to patients who do not respond to TNF-α inhibitors. 43
Spontaneous (Including Genetically Engineered) Models
A number of inbred mouse strains are prone to develop arthritis. Such strains are particularly useful for the identification of genes that contribute to the pathogenesis of autoimmunity. For example, a whole-genome map for traits associated with spontaneous arthritis has been derived from analysis of arthritic BXD2/TyJ mice. 45 Because susceptibility to arthritis in strains such as the BXD2 is polygenic, the individual variability of timing and extent of lesions is greater in these strains than in robust inducible models. Therefore, such strains are not commonly used for studies that address efficacy of specific therapies or explore specific mechanisms of disease.
In contrast to reliance on polygenic traits, targeted genetic engineering enables more narrowly defined and less complex spontaneous disease models. For RA, such models typically aim to either suppress key anti-inflammatory pathways or exaggerate key proinflammatory pathways. 15 An important, representative example of the first strategy is the IL-1 receptor antagonist (IL-1RA) knockout mouse. IL-1 is a proinflammatory cytokine produced by a variety of cell types, including activated monocytes, macrophages, fibroblasts, and synovial cells—all of which are important contributors to joint inflammation and destruction in RA. 10 The naturally occurring IL-1 inhibitor IL-1RA is critical for the limitation of excessive inflammatory responses. The relevance of IL1-RA has been clinically validated by the approval of recombinant human IL-1RA as a biotherapeutic (anakinra, Kineret, Amgen). IL-1RA-null mutant (knockout) mice backcrossed onto a BALB/c background spontaneously develop arthritis 19 with lesions that closely resemble RA, including proliferation of synovial lining cells, pannus formation, osteolysis, and cartilage erosion. 19 In addition to its phenotypic similarity to human RA, this knockout mouse model shares other features with RA, such as the presence of rheumatoid factor—autoantibodies to double-stranded DNA and to type II collagen. 19 IL-6 and TNF-α are increased in arthritic joints, and IL-17 is required for arthritis in this model. 18,25,36 T cells are important in the pathogenesis of human RA 8,30,49,55 and are central to the pathogenesis of arthritis in IL-1RA knockout mice: Whereas T cell–deficient IL-1RA knockout mice do not develop arthritis, T-cell transfer from IL-1RA knockout mice to (T-cell deficient) nude mice induces arthritis. 18,36
The second strategy—that is, to generate overactivity of critical proinflammatory mediators—is best exemplified by models that are driven by TNF-α. The TNFΔ ARE mouse was created by deletion of the AU-rich elements (AREs), which mediate rapid degradation of TNF-α transcripts unless the transcripts are stabilized during cell activation. 5 Because TNF-α is primarily regulated at the posttranscriptional level, ARE loss results in transcript stabilization and persistent TNF-α elevation. TNFΔ ARE mice 26 develop arthritis, transmural inflammation of the small intestine, and growth retardation. After rapid onset, the disease is progressive and fatal between 5 and 12 weeks of age in homozygous mice. The disease course is more protracted in heterozygous or hemizygous animals, which show overt arthritis at age 6 to 8 weeks and are therefore more commonly used in experimental studies. By 16 weeks, polyarthritis in heterozygous TNFΔ ARE mice involves nearly all the paw joints usually examined. Lesions are characterized by extensive fibroplasia, cartilage injury, and osteolysis. Because there is only minimal formation of new bone, ankylosis is not a feature of this model in spite of severe osteolysis (Figs. 12–14). The lack of new bone formation and the growth retardation also seen in these mice are likely attributable to compromised calcium homeostasis with reduced intestinal calcium absorption. 20 The TNFΔ ARE model (like other similar TNF-α-driven models) is useful to gauge therapies for their benefit against reference treatment with a TNF-α inhibitor, which, depending on treatment onset and regimen, substantially attenuates lesions.
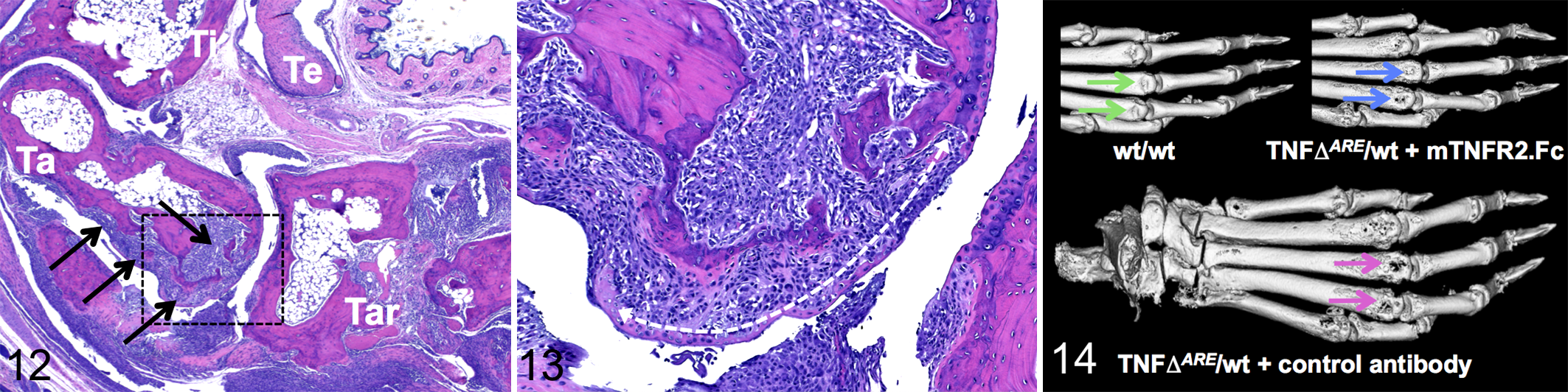
In summary, this review provides a synopsis of the more commonly used mouse models of autoimmune arthritis with an emphasis on the underlying biological mechanisms particular to the individual models. Even though overlapping mechanisms in the effector phase account for a remarkably uniform morphology of lesions across the models, care must be taken in the selection of models and interpretation of findings since some of the initial mechanisms or pathways may not be shared among the models.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.