
Abstract
Traditionally, control of phosphorus in the body has been considered secondary to the tighter control of calcium by parathyroid hormone and vitamin D. However, over the past decade, substantial advances have been made in understanding the control of phosphorus by the so-called phosphatonin system, the lynchpin of which is fibroblast growth factor 23 (FGF23). FGF23 binds to the klotho/FGFR1c receptor complex in renal tubular epithelial cells, leading to upregulation of Na/Pi cotransporters and subsequent excretion of phosphorus from the body. In addition, FGF23 inhibits parathyroid hormone and the renal 1α-hydroxylase enzyme, while it stimulates 24-hydroxylase, leading to decreased 1,25-dihydroxyvitamin D3. FGF23 is intimately involved in the pathogenesis of a number of diseases, particularly the hereditary hypophosphatemic rickets group and chronic kidney disease, and is a target for the development of new treatments in human medicine. Little work has been done on FGF23 or the other phosphatonins in veterinary medicine, but increases in FGF23 are seen with chronic kidney disease in cats, and increased FGF23 expression has been found in soft tissue sarcomas in dogs.
Keywords
Fibroblast growth factor 23 (FGF23) is a recently recognized hormone that plays an intrinsic role in calcium and phosphorus metabolism, both in health and disease. Studies have shown that in humans, FGF23 is implicated in multiple inherited bone diseases, paraneoplastic bone disease (tumor-induced osteomalacia), and complications of chronic kidney disease and cardiovascular disease. In animals, a handful of studies have already recognized potential roles for FGF23 as a biomarker of disease and perhaps as a therapeutic agent. This review details the manner in which FGF23 joins established players in calcium-phosphorus metabolism, current knowledge of its role in disease in humans and animals, and future avenues for research and use of this hormone.
Traditional Model of Calcium-Phosphorus Metabolism
Our understanding of calcium-phosphorus metabolism originated in the study of rickets, one of the first diseases to be investigated scientifically. 21,188 An increase in the incidence of rickets occurred in Europe during the Industrial Revolution, associated with migration of people into cities, a marked increase in atmospheric pollution, overcrowding, and poor diet. 148 It was deduced that exposure to sunlight and cod liver oil prevented rickets, and researchers eventually identified the preventative substance within cod liver oil as vitamin D3. 21,148,188
Further research eventually defined the critical role vitamin D3 plays in managing plasma calcium and phosphorus concentrations and the related roles of important hormonal messengers such as parathyroid hormone (PTH). This led to the classic model of calcium-phosphorus metabolism (Fig. 1). Briefly, changes in plasma calcium concentration are detected by calcium-sensing receptors (CaSR) in the parathyroid gland, which modulate the release of PTH. 46 PTH increases plasma calcium concentrations by increasing osteoclastic resorption of bone (via osteoblastic stimulation), increasing tubular reabsorption of calcium in the kidney, and increasing active vitamin D3 production. 24 PTH also downregulates tubular reabsorption of phosphorus. 186
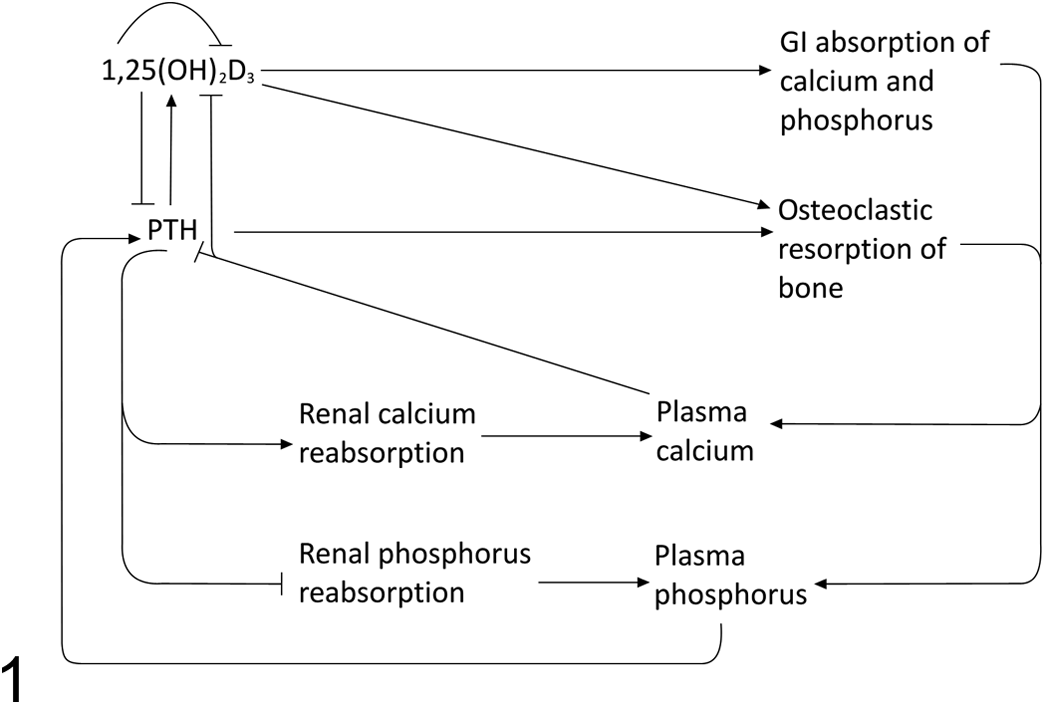
Classic model of calcium, phosphorus, vitamin D, and parathyroid hormone metabolism (arrow = upregulates/increases, bar = downregulates/decreases). Active vitamin D (1,25(OH)2D3) increases absorption of calcium and phosphorus from the gastrointestinal (GI) tract, increases the release of calcium and phosphorus from bone by osteoclasts, and inhibits the production of parathyroid hormone (PTH) by the parathyroid gland. PTH increases calcium reabsorption from the kidney but decreases phosphorus reabsorption; it also increases the release of calcium and phosphorus from bone by osteoclasts.
Dietary or ultraviolet light–induced cutaneous vitamin D precursors must be activated to become functional. A hydroxyl group is added in the liver to form 25-hydroxyvitamin D3 (25OHD3), followed by a second hydroxylation in the kidney by the renal 25-hydroxyvitamin D3-1α-hydroxylase enzyme (1α-hydroxylase) to produce active vitamin D3 (1,25(OH)2D3). 78 Active vitamin D3 then acts on the gastrointestinal tract to upregulate absorption of calcium and phosphorus and on bone to increase osteoclastic resorption. It may have direct effects on renal calcium and phosphate reabsorption and downregulates PTH and 1α-hydroxylase activity. 23,46,62,135 If dietary vitamin D3 or exposure to ultraviolet radiation is inadequate, insufficient 25OHD3 is available for conversion to 1,25(OH)2D3, resulting in low plasma calcium and phosphorus, both of which are required for bone mineralization. Hyperparathyroidism may then develop as a consequence of hypocalcemia.
Control of phosphorus metabolism was once thought to be indirect rather than under the primary control of any one hormone. 94 Parathyroid hormone prevents renal tubular phosphate reabsorption, and vitamin D3 increases phosphorus absorption from the gastrointestinal tract; however, it was believed that these processes were related to calcium homeostasis. This model was revisited when investigation of X-linked hypophosphatemic rickets (Hyp) in mice suggested the existence of a circulating factor that reduced the activity of renal proximal tubular cell Na/Pi cotransporters and suppressed activation of 25OHD3. This phosphate-regulating substance was tentatively named phosphatonin, 47 and over time it became clear that multiple previously unsuspected physiological pathways influenced phosphorus metabolism in health and disease.
Fibroblast Growth Factor 23—Physiology in Humans and Rodent Models
In 2000, a novel secreted member of the fibroblast growth factor family of mitogens was identified. 218 Just prior to this discovery, another group of researchers found that mutations in the same protein, named fibroblast growth factor 23 (FGF23), were associated with autosomal dominant hypophosphatemic rickets. 3 Studies in mice and rats indicated that expression of FGF23 in bone or teeth was higher or more easily detected than in other tissues, 113,115,224 and other studies confirmed that osteoblasts and osteocytes are the main physiological source of FGF23. 96,102,132
Various researchers established that FGF23 causes renal phosphate wasting, reduces gastrointestinal phosphate absorption, and reduces activation of vitamin D3 (Fig. 2). Chinese hamster ovary cells expressing human FGF23, when transplanted into nude mice, caused hypophosphatemia, phosphaturia, high serum alkaline phosphatase, low 1,25 (OH)2 vitamin D3 (secondary to reduced 1α-hydroxylase expression), bone deformity, and reduced weight gain with histological findings consistent with rickets. 173 Similarly, transgenic mice expressing human FGF23 had renal phosphate wasting and osteomalacia secondary to reduced expression of type I, IIa, and IIc renal proximal tubular Na/P cotransporters (Npt) and reduced activation of vitamin D3, 102,176 and intravenous administration of recombinant human FGF23 to mice led to hypophosphatemia and reduced 1,25(OH)2 vitamin D3 concentrations 171 —thus, the FGF23 results in phosphaturia, hypophosphatemia, and decreased plasma 1,25(OH)2D3 concentrations.
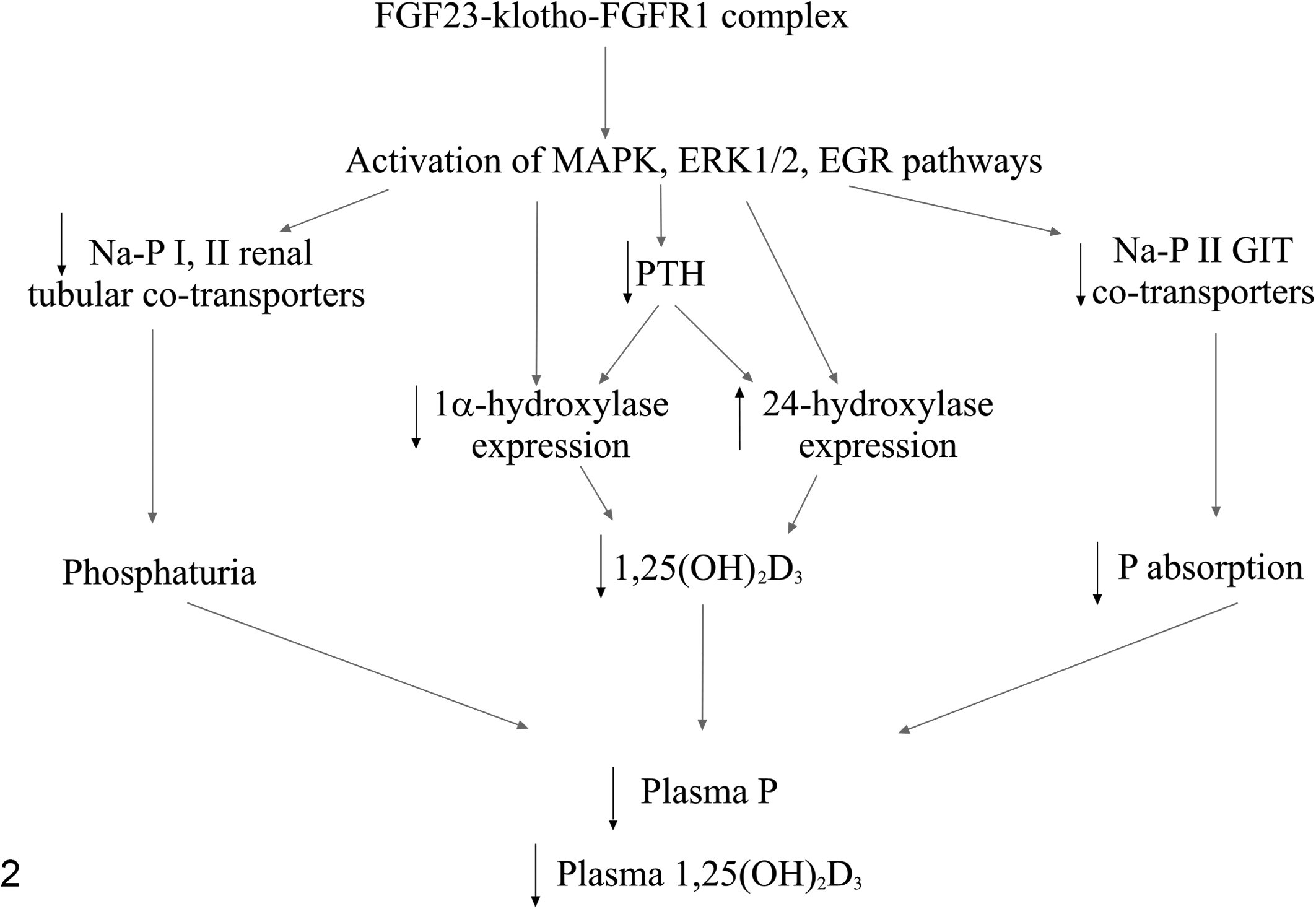
Functions of fibroblast growth factor 23 (FGF23) and FGF23 signaling pathways. The FGF23-klotho-fibroblast growth factor receptor 1 (FGFR1) complex activates the mitogen-activated protein kinase (MAPK) and extracellular signal-related kinase-1/2 (ERK1/2) and early growth response (EGR) signaling pathways. This results in a decrease in Na/P renal and gastrointestinal tract (GIT) cotransporters, leading to phosphaturia and decreased GIT phosphate (P) absorption, respectively. At the same time, plasma PTH concentrations decrease, leading to decreased expression of renal 1α-hydroxylase and increased expression of 24-hydroxylase, resulting in decreased plasma 1,25(OH)2D3 concentrations. The overall result of activation of the FGF23 signaling pathway is a decrease in plasma phosphorus concentration and decreased plasma 1,25(OH)2D3 concentration.
FGF23 binds fibroblast growth factor receptors (FGFRs), predominantly FGFR1c, although binding to others (2c, 3c, and 4) has been demonstrated. 109,116,217,225 It has also been suggested that there is a renal tubular basolateral membrane receptor for FGF23, which is functionally distinct from the known FGFR family. 220 While FGFRs are widely expressed throughout the body, effective FGFR binding of FGF23 requires the presence of its cofactor, αKlotho, 81,100 a transmembrane or secreted glycoprotein that is predominantly expressed on renal distal tubular epithelial cells, 110 on proximal renal tubular epithelial cells at a lower concentration, 79,146 and in the parathyroid gland. 18 Cell surface molecules such as heparin and glycosaminoglycans may help to stabilize the FGF23–Klotho–FGFR1(IIIc) receptor complex. 194
Klotho –/– mice have a very similar phenotype to Fgf23 –/– mice, 110,192,223 and Klotho expression levels are inversely related to levels of Fgf23 expression. 124 Fgf23 –/–/Klotho –/– mice have a similar phenotype to knockout mice of either gene, and administration of Fgf23 does not rescue their phenotype, confirming that Fgf23 requires Klotho to alter systemic phosphorus homeostasis. 137 The role of secreted Klotho as a hormone is currently unclear, 81 but it has been suggested that circulating Klotho could protect FGF23 from degradation, 25 and secreted Klotho may have FGF23-independent phosphaturic effects. 79
After forming the FGF23-Klotho-FGFR1c complex on renal tubular epithelial cells and the parathyroid gland, FGF23 initiates mitogen-activated protein kinase (MAPK) and extracellular signal-related kinase-1/2 (ERK-1/2) or early growth response-1 (EGR-1) pathways (Fig. 2). 18,149,217 These signals lead to phosphorylation of the Na+/H+ exchange regulatory cofactor (NHERF) 1, which causes existing cotransporters to be removed from the brush-border membrane of renal tubular epithelial cells, and reduced expression of renal tubular Na/P II cotransporters. 6,139 In addition, the ERK-1/2 pathway (induced by FGF23) downregulates renal 1α-hydroxylase expression. 146 FGF23 upregulates renal 24-hydroxylase and downregulates intestinal Na/P cotransporters in a vitamin D3 receptor-dependent manner 86 but may also modulate 24-hydroxylase and 1α-hydroxylase activity via its effects on PTH 11,13 as FGF23 directly downregulates PTH in an ERK-1/2–dependent manner via the MAPK pathway (Fig. 2). 18,97,177 In 1 study, FGF23 also increased expression of the parathyroid calcium-sensing receptor and vitamin D receptor, while decreasing parathyroid cell proliferation. 28 Interestingly, another study found that PTH concentrations decreased in mice when FGF23 was neutralized with antibodies, suggesting that increased concentrations of 1,25(OH)2 vitamin D3 in these mice were more important in modulating PTH concentrations. 219 Therefore, increased FGF23 signaling results in decreased 1α-hydroxylase activity and increased 24-hydroxylase activity, leading to decreased plasma 1,25(OH)2D3, and decreased renal Na/P cotransporters, resulting in phosphaturia and hypophosphatemia (Fig. 2).
Crosstalk between FGF23 signaling pathways and other systems has been detected. Proximal tubule phosphate transport can be responsive to prostaglandin E2 (PGE2), and FGF23 may influence renal PGE2 concentrations through the MAPK pathway. 16,181 Members of the Wnt signaling system may be expressed in the kidney in response to FGF23. 52 Fgf23 –/– mice show some abnormalities unrelated to mineral metabolism, including atrophy of the thymus and spleen, low serum triglycerides, increased serum cholesterol, and hypoglycemia, 172 with increased peripheral insulin sensitivity and high subcutaneous glucose tolerance. 77 Leptin has been shown to stimulate FGF23 expression. 191 These findings suggest a relationship between FGF23 and energy metabolism. There is currently uncertainty over the relationship between estrogen and FGF23. 205
FGF23 may also act as a paracrine messenger within bone. 179 When FGF23 was overexpressed in osteoblast cell culture, formation of osteoid nodules and matrix mineralization were inhibited. 197 While some researchers did not find any evidence of Klotho expression in bone, 116 others 150 showed that Klotho and FGFR1 are present in the osteocytes and osteoblasts of DMP1-caPTHR1 transgenic mice (which have constitutive upregulation of PTH signaling in osteocytes) and observed elevations in early growth response genes 1 and 2 (EGR-1/2), consistent with FGF23 signaling in these cells. 150
Regulation of FGF23 is mediated through several mechanisms, including 1,25(OH)2D3, calcium, phosphate, and PTH. There is general agreement that FGF23 is upregulated by active vitamin D3; 33,88,114,162,171 for example serum FGF23 concentration is increased in rodents given calcitriol. 96 Physeal chondrocytes of growing mice receive the 1,25(OH)2 D3 message and then secrete a substance to control FGF23 production in bone. 126
Despite some conflicting evidence and the difficulty of separating the effects of calcium on FGF23 from the effects of PTH and 1,25(OH)2 vitamin D3, it is thought that plasma calcium concentrations play a role in FGF23 regulation. This has been demonstrated in a carefully controlled trial, 36 and given the downregulatory effects of FGF23 on PTH and 1,25(OH)2 vitamin D3, a corresponding upregulatory effect of calcium on FGF23 completes a feedback loop. Supporting evidence includes studies where plasma FGF23 concentrations increased in vitamin D3 receptor knockout mice and parathyroidectomized rats given a calcium loading diet, 152,175 FGF23 concentrations decreased in parallel with the rate of calcium (not PTH) decline after parathyroidectomy in patients with primary hyperparathyroidism, 95 and FGF23 concentrations decreased when rats were fed a diet low in calcium and vitamin D. 152 It seems that normocalcemia might be required to permit PTH and 1,25(OH)2 vitamin D3 regulation of FGF23. It is noted that the effects of calcium on FGF23 may be long term rather than short term, since acute changes in plasma calcium do not seem to alter plasma FGF23 concentrations. 202 Independent of vitamin D, FGF23 appears to also have a role in the regulation of renal calcium reabsorption. 5 In contrast to the inhibitory effects of FGF23 on the production of 1,25(OH)2D3, FGF23 seems to stimulate calcium reabsorption in the kidney via the TRPV5 (transient receptor potential vanilloid 5) calcium channel, thus minimizing any effects on plasma calcium concentration. 5
While it has been shown that phosphate regulates FGF23 expression by cultured osteoblasts in vitro, 132 and phosphate increases FGF23 independently of 1,25(OH)2 vitamin D3, 226 in vivo studies examining the response of FGF23 to dietary or plasma phosphate have produced apparently conflicting results. FGF23 was increased in hyperphosphatemic humans with hypoparathyroidism 71 and in hyperphosphatemic Hyp mice (model of X-linked hypophosphatemic rickets) with a Pth deletion. 12 FGF23 concentrations respond to dietary phosphate in normal humans, normal mice, Npt2 –/– (sodium/phosphorus co-transporter 2) mice (although at a lower level), and Hyp mice. 27,56,136,145,162 A recent study in rats treated with oral phosphorus, a calcium-sensing receptor activator, and injectable PTH suggested that the increase in FGF23 may be triggered by increased PTH. 182 Conversely, FGF23 did not increase in tandem with serum phosphate in men with hypogonadism, 26 acute dietary phosphate loading did not increase FGF23 in healthy volunteers, 103 and acute phosphate infusion did not alter FGF23. 89 The discrepancies in these studies regarding phosphorus and FGF23 concentrations may relate to how phosphate is sensed (still currently unknown) 81 and what FGF23 is actually regulating (eg, plasma phosphate vs the local phosphate concentration near osteocytes, vitamin D3, or bone metabolism). 211 It has been proposed that FGF23 increases in response to long-term rather than short-term increases in plasma phosphate. 103
Whether PTH had an effect on FGF23 production by bone was unclear, again due to the confounding effects of other players in calcium-phosphorus metabolism. 201,215 For example, parathyroidectomy in renal secondary hyperparathyroidism leads to a reduction in FGF23 concentrations, 164 but significantly increased FGF23 has not been found in many cases of primary hyperparathyroidism. 178,184 However, it is now generally accepted that PTH directly stimulates FGF23 production, 105,150 and recent evidence suggests that it achieves this via the Nurr1 (nuclear receptor-related 1 protein) receptor. 128 It may also indirectly increase FGF23 concentrations through its effects on 1,25(OH)2 vitamin D3 and its effects on paracrine bone factors. 36
In summary, 1,25(OH)2D3, PTH, and long-term increases in calcium or phosphorus lead to increased FGF23 concentrations. Investigation of the role of FGF23 in disease led to our current understanding of phosphorus metabolism, as presented in Figure 3, which involves several subsidiary players described in more depth below.
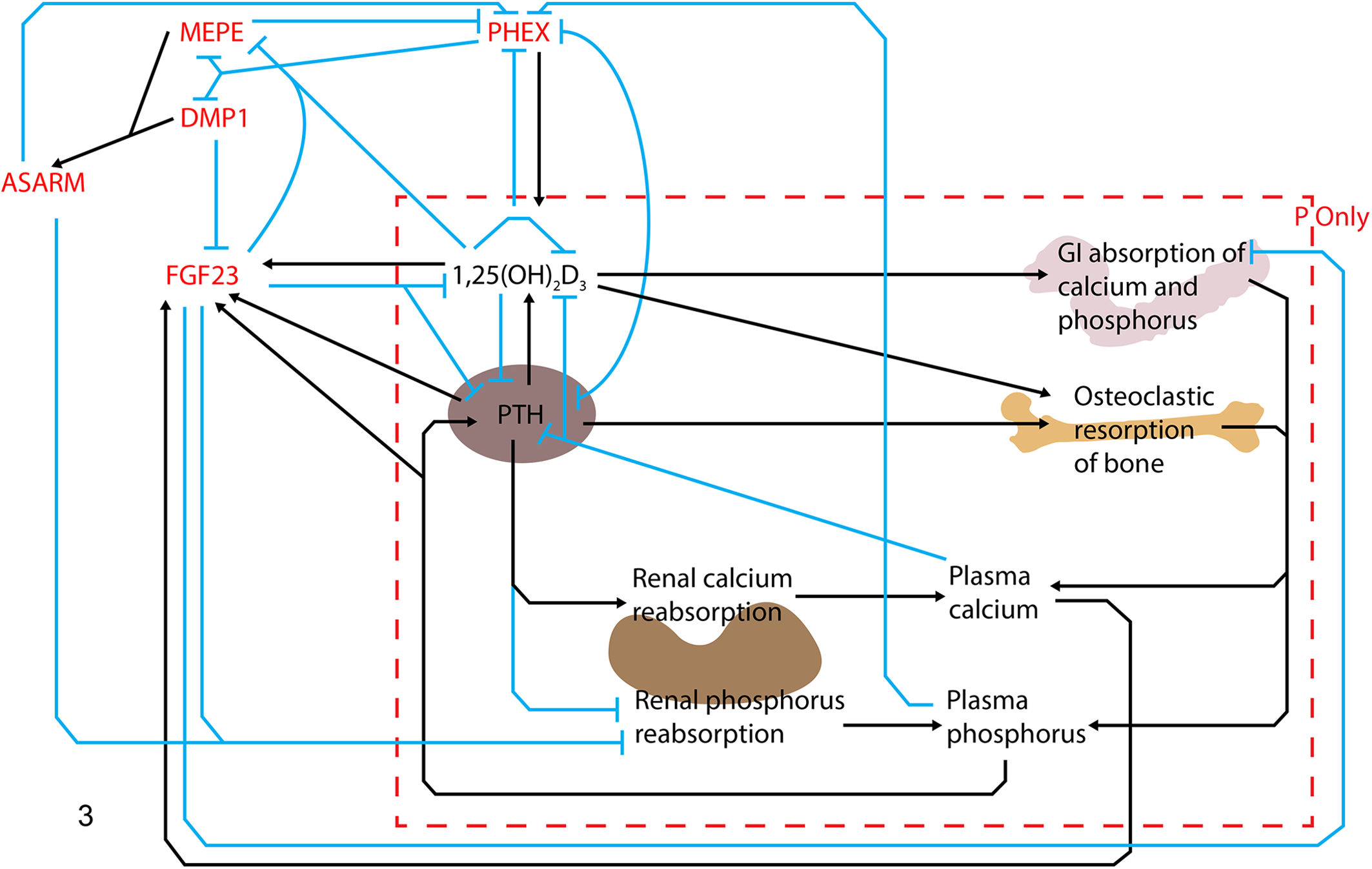
A summary of the main players in phosphorus (P) metabolism. Minor regulatory pathways and hormones or factors peripheral to calcium-phosphorus control are not depicted (arrow = upregulates/increases, bar = downregulates/decreases). The central player in phosphorus metabolism is fibroblast growth factor 23 (FGF23), and it inhibits renal phosphorus reabsorption, decreases active vitamin D (1,25(OH)2D3) production, and inhibits production of parathyroid hormone (PTH) by the parathyroid gland. Plasma FGF23 concentrations are increased by 1,25(OH)2D3, PTH, and long-term hypercalcemia and hyperphosphatemia, and are regulated by other components of the phosphatonin pathways including matrix extracellular phosphoglycoprotein (MEPE), dentin matrix protein 1 (DMP1), phosphate-regulating gene with homology to endopeptidases on the X chromosome (PHEX), and acidic serine–aspartate-rich MEPE associated motif (ASARM). GI, gastrointestinal. The authors would like to acknowledge the artistic effort of Kyle Brown on this figure.
Fibroblast Growth Factor 23—Disease in Humans and Lessons From Mouse Models
It is through investigating the different types of inherited hypophosphatemic rickets, particularly autosomal dominant hypophosphatemic rickets, that researchers have been able to unravel (for the most part) the pathways involved in FGF23-mediated control of phosphorus and the other compounds involved in bone mineralization. 3
Rickets and osteomalacia are syndromes of deficient calcium and phosphorus deposition in osteoid during bone synthesis. 42 Failure of normal mineralization in the physeal or articular cartilage of children leads to rickets, characterized by thickening and flaring of physes, bowing of long bones, bone and joint pain, delayed growth, delayed standing or walking, frequent falling, and even seizures due to hypocalcemia. 188,206 Osteomalacia is observed in adults and leads to more occult signs such as bone pain, gait instability, muscle weakness, and pathological fractures. 188
A consortium of researchers identified FGF23 mutations in families affected by autosomal dominant hypophosphatemic rickets (ADHR), 3 and experimental evidence suggested that the mutated FGF23 in human patients with ADHR resists posttranslational processing, leading to excessive FGF23 signaling. 22,174,207 Rats or mice injected with the ADHR FGF23 mutant have reduced expression of renal Na/P cotransporters IIa and IIc, 166 reduced activity and abundance of intestinal brush-border membrane type IIb intestinal Na/P cotransporters, 133 and reduced expression of renal 1α-hydroxylase. Thus, the excessive FGF23 signaling in patients with ADHR results in phosphaturia, hypophosphatemia, and inappropriately low serum 1,25(OH)2D3 concentrations, the consequence of which is the development of rickets (Fig. 4). 161
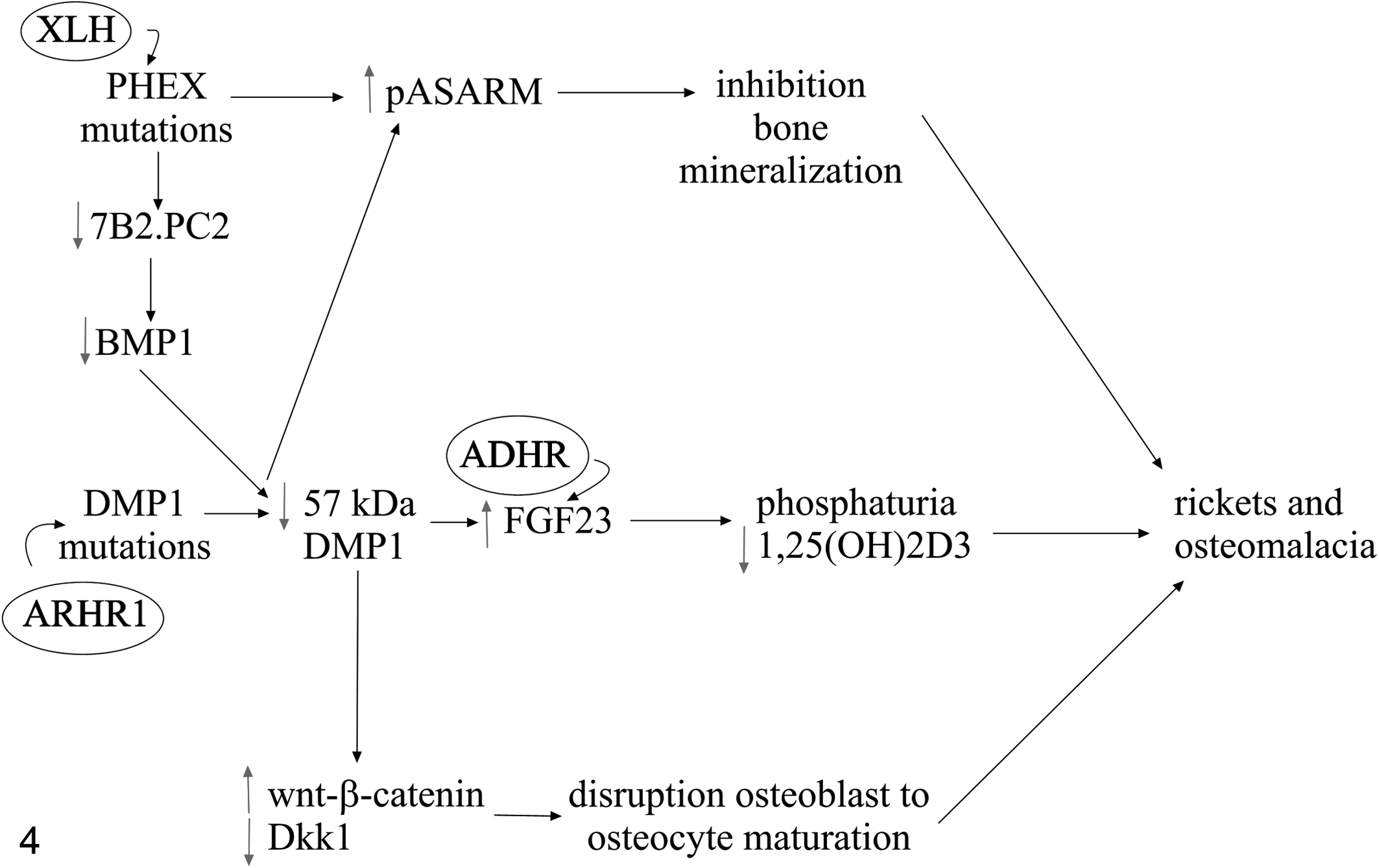
Pathogenesis of the genetic mutations involved in 3 forms of hypophosphatemic rickets in humans. X-linked hypophosphatemic rickets (XLH) is due to PHEX (phosphate regulating gene with homology to endopeptidases on the X chromosome) mutations, which result in decreased levels of the 7B2.PC2 (proprotein convertase and its chaperone) heterodimer, leading to decreased activation of bone morphogenic protein 1 (BMP1) and subsequently decreased formation of the active 57-kDa fragment of dentin matrix protein 1 (DMP1). Mutations in DMP1 cause autosomal recessive hypophosphatemic rickets type 1 (ARHR-1), again leading to decreased levels of the 57-kDa fragment of DMP1. This results in increased FGF23 (fibroblast growth factor 23), which also occurs in humans with autosomal dominant hypophosphatemic rickets (ADHR), in whom there is a defect in posttranslational processing of FGF23. Increased plasma FGF23 concentrations lead to phosphaturia and decreased plasma 1,25(OH)2D3 concentrations. PHEX and DMP1 mutations also result in increased levels of phosphorylated acidic serine-aspartate rich MEPE associated motif (pASARM), which inhibits bone mineralization. In addition, decreased levels of 57-kDa DMP1 lead to increased signaling in the wnt-β-catenin pathway, with decreased Dkk1 (Dickkopf 1), resulting in disruption of osteoblast to osteocyte maturation. Consequently, the inhibition of bone mineralization, phosphaturia, decreased 1,25(OH)2D3, and disruption of osteoblast to osteocyte maturation result in the rickets and osteomalacia seen clinically in affected patients.
A mirror image of this disease, tumoral calcinosis, was found to be related to a lack of FGF23, leading to hyperphosphatemia, hypophosphaturia, normal to increased vitamin D3 concentrations, and generalized soft tissue mineralization. 123 Defective FGF23 activity in this disease can be due to a mutated FGF23 gene causing a truncated protein, 7 low secretion of intact FGF23, 19 secretion of a rapidly degraded FGF23 mutant, 101,104 mutation of klotho, 83 or inadequate posttranslational processing by UDP-N-acetyl-α-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3 (ppGalNacT3) or an intermediary. 189 Supporting this, the Fgf23 –/– mouse model has a short life span, growth retardation, hyperphosphatemia due to increased renal proximal tubular phosphate reabsorption, and increased 1,25(OH)2 vitamin D3 secondary to high 1α-hydroxylase activity. 172
There are a number of different forms of hypophosphatemic rickets, all related to defects in proteins that regulate FGF23 signaling (Fig. 4), and investigation of these diseases has assisted with elucidating the regulation of FGF23. Human families with autosomal recessive hypophosphatamic rickets type 1 (ARHR-1) have mutations in dentin matrix protein 1 (DMP1). 55,118 These patients have renal phosphate wasting, inappropriately low 1,25(OH)2D3, and increased FGF23 levels, leading to excessive deposition of hypomineralized osteoid. Dentin matrix protein 1 (DMP1) is a member of the SIBLING (small integrin-binding ligand, N-linked glycoproteins) family and has been shown to modulate postnatal dentinogenesis, 221 chondrogenesis, and osteogenesis. 222 SIBLINGs are characterized by a highly conserved C-terminal acidic serine–aspartate-rich MEPE-associated motif (ASARM), which can be released after cleavage by Zn metallopeptidases such as bone morphogenetic protein 1 (BMP1), and PHEX (phosphate-regulating gene with homology to endopeptidases on the X chromosome). 1,2,37,153
The functional domain of DMP1 is a 57-kDa C-terminal fragment, 121 and it regulates production of FGF23 by blocking transcription of FGF23 messenger RNA (mRNA), 107,125,213 and thus the increased plasma FGF23 concentration results in hypophosphatemia and impaired bone mineralization. In addition, DMP1 is also directly involved in bone mineralization; mutations in DMP1 result in increased wnt/β-catenin signaling and decreased Dkk1 (Dickkopf 1) 29 with disruption of normal osteoblast to osteocyte maturation. 229 Mutations in DMP1 therefore cause ARHR-1 by increasing plasma FGF23 concentrations, resulting in phosphaturia, hypophosphatemia, and decreased plasma 1,25(OH)2D3, in addition to interfering with the maturation of osteocytes; as a consequence, affected individuals develop rickets (Fig. 4).
X-linked hypophosphatemic rickets (XLH) is a disease of humans characterized by shortened stature, vitamin D3 nonresponsive rickets or osteomalacia, hypophosphatemia, and phosphaturia 68 modeled by the Hyp mouse. 49 The rachitic changes result from renal phosphate wasting and hypophosphatemia 159 combined with inappropriately low vitamin D3 activation, secondary to inadequate activation of renal 1α-hydroxylase 117,122,129 and excessive 24-hydroxylation of 1,25-(OH)2 D3 or 25(OH) D3. 35,185,187 Mutations responsible for the XLH phenotype have been mapped to a human X chromosome region encoding PHEX. 82 Phex mutations seem to prevent export of PHEX to the cell membrane, 158 leading to altered osteocyte proliferation and differentiation, altered expression of noncollagenous bone proteins, 130,131 and defective mineralization. 170,214
PHEX/Phex is expressed exclusively on the border of osteoblasts and odontoblasts of humans and normal embryonic/postnatal/adult mice 17,70,155 and promotes matrix mineralization. Individuals with XLH have markedly increased serum FGF23 concentrations, but it is only recently that the mechanism relating PHEX mutations to FGF23 dysregulation has been determined. FGF23 production and breakdown in osteocytes/osteoblasts are controlled by the 7B2.PC2 heterodimer. 227 PC2 is a proprotein convertase, while 7B2 is its chaperone. 196 Decreased levels of functional PHEX protein result in decreased 7B2.PC2 heterodimer levels, leading to decreased BMP1 and impaired breakdown of DMP1 into its active 57-kDa C-terminal fragment, and this in turn increases transcription of FGF23 mRNA (Fig. 4). 54,227 The bone mineralization defect is likely due to 2 processes: decreased 57-kDa C-terminal DMP1, resulting in increased wnt/β-catenin signaling, and decreased Dkk1, as occurs in ARHR-1 with DMP1 mutations. 54,120,121
PHEX also degrades a number of extracellular matrix proteins, including osteopontin and matrix extracellular phosphoglycoprotein (MEPE), which belong to the SIBLING family of extracellular matrix glycoproteins. 2,15,154 The phosphorylated ASARM peptide of MEPE and osteopontin inhibits mineralization (Fig. 4) and may also directly inhibit renal phosphate transport. 1,2,15,37,228
Fibrous dysplasia and McCune-Albright syndrome (MAS) are diseases of humans featuring somatic mutations in GNAS1 (guanine nucleotide-binding protein, α-stimulating activity polypeptide 1), which encodes the α subunit of the stimulatory G protein (Gs), leading to constitutive activation of adenylyl cyclase. 165 A small number of affected patients have renal phosphate wasting, have inappropriately low levels of active vitamin D3, and develop rickets or osteomalacia. 32 FGF23 was found to be expressed by cells of osteogenic origin within regions of fibrous dysplasia, and increased FGF23 in affected patients correlated with phosphate wasting and level of disease burden (ie, number of mutated cells). 151
In 2010, a research group reported a novel mutation of the ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) enzyme responsible for forming pyrophosphate (a mineralization inhibitor). Mutations in this gene cause both autosomal recessive hypophosphatemic rickets type 2 (ARHR-2) and generalized arterial calcification of infancy. 119 The mechanism by which ENPP1 mutations can cause such disparate clinical syndromes is unknown. Plasma FGF23 concentrations are mildly increased or inappropriately normal in patients with ARHR-2. 108,119
Tumor-induced osteomalacia (TIO) is a rare paraneoplastic syndrome seen in a small number of human patients, first reported in 1947. 127 The usual history is one of chronic bone or muscle pain, fatigue, muscle weakness and lethargy, gait abnormalities, height loss, or poor growth, sometimes leading to pathological fractures. 156,198 The responsible tumors are usually small and located in obscure areas, such as the paranasal sinuses 200 or within bone, 9,156 and are generally difficult to diagnose and resect.
Although occasionally reported in association with carcinomas 156,195 or linear nevus sebaceous syndrome, 8,30 most tumors causing TIO are designated “phosphaturic mesenchymal tumor (mixed connective tissue variant)” or PMT-MCT. 199 The tumors have a consistent morphologic appearance of loosely to densely packed spindle- to stellate-shaped cells embedded within a highly vascular myxoid to chondroid stroma, which is frequently described as “grungy” due to diffuse mineralization. Adipocytes, hemangiopericytoma-like vessels lacking “staghorn” branching, osteoclast-like multinucleated giant cells, microcystic change, local infiltration, hemorrhage, and osteoid formation are also frequently reported. 51,60,198,199
Patients with tumor-induced osteomalacia show reduced renal phosphate reabsorption, leading to hypophosphatemia, inappropriately low concentrations of 1,25(OH)2D3, and osteomalacia 45,63,156 producing a phenotype similar to XLH and ADHR. Removal of the causative tumor leads to resolution of the osteomalacia. 51,147,156,180 FGF23 protein and mRNA are present and overexpressed in the tumor cells of patients with TIO. In addition, increased serum concentrations of FGF23 are often detected in patients with TIO by enzyme-linked immunosorbent assay (ELISA), and these quickly return to normal after the causative tumors are removed. 74,85,93,183 Other phosphatonins, such as MEPE, secreted frizzled related protein 4 (SFRP4), and DMP1, are often coexpressed with FGF23 in TIO, 38,74,84,190 and PTH or a PTH-like substance may also play a role in the pathogenesis of TIO. 92,99,138,167,169
FGF23 is intimately associated with the pathogenesis of chronic kidney disease (Fig. 5), 81 now known in human medicine as chronic kidney disease–mineral and bone disorder (CKD-MBD). 134 Increases in serum FGF23 occur in parallel with the stage of kidney disease in human patients, and increases in FGF23 are a risk factor for progression of disease. 59,74 It was initially thought (and still may be the case) that increases in FGF23 were the result of impaired phosphorus excretion as the kidney failed (see also recent reviews on renal phosphorus handling 67,106 ); however, increases in FGF23 occur prior to any increases in PTH or hyperphosphatemia. 87,103 In addition, research has shown that in humans with normophosphatemic mild chronic kidney disease, serum FGF23 concentrations increase as glomerular filtration rate declines. 10,57,203 Although serum phosphorus concentrations also increase with decreasing glomerular filtration rate, the increase in serum phosphorus appears to occur after the increase in serum FGF23. 31 However, as kidney disease becomes more severe, serum phosphorus concentrations may be a more significant predictor of serum FGF23 concentrations. 203
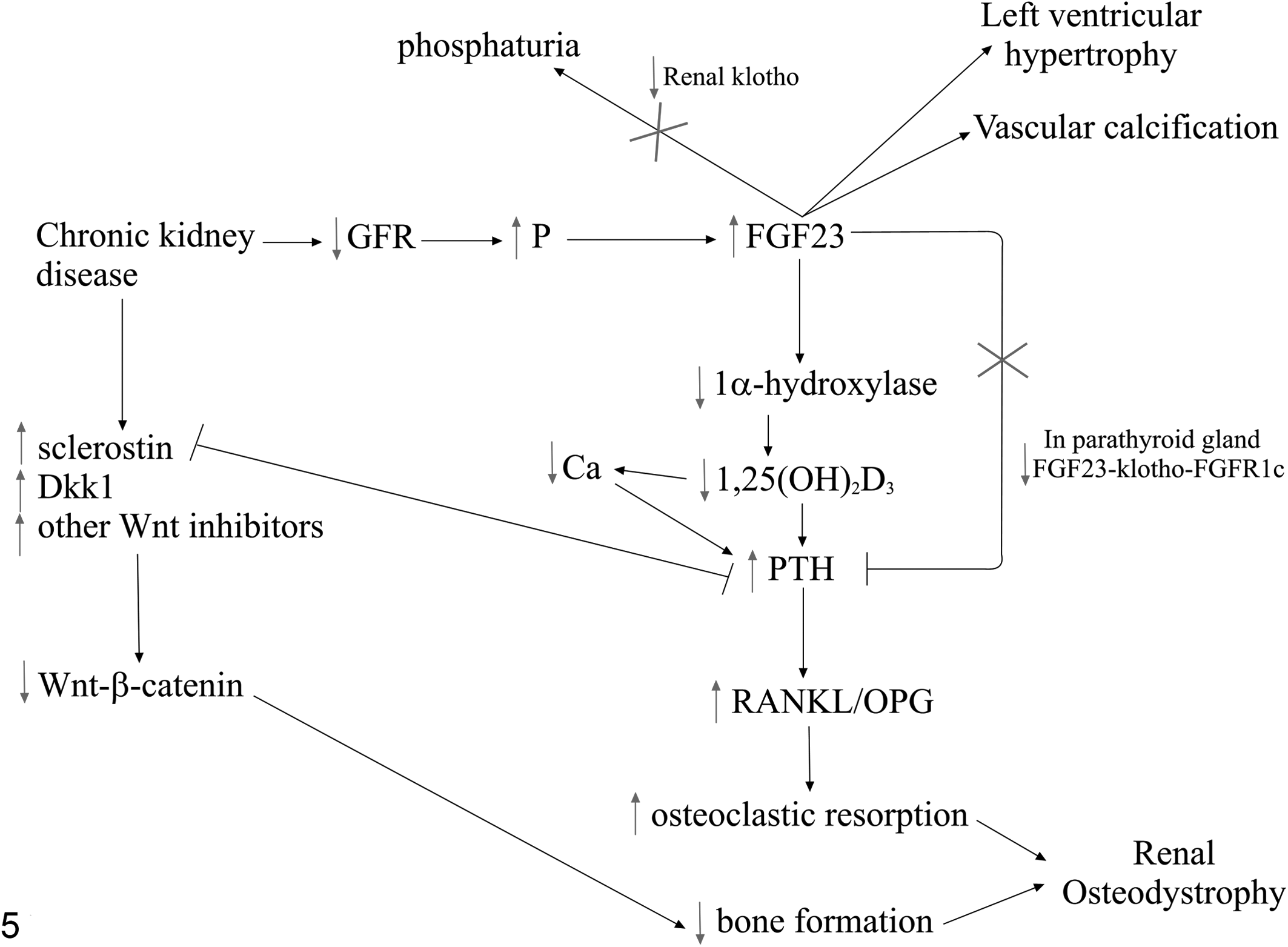
Pathogenesis of fibroblast growth factor 23 (FGF23) and chronic kidney disease. Plasma FGF23 concentrations increase early in chronic kidney disease, although it is unclear whether decreased glomerular filtration rate (GFR), hyperphosphatemia, or increased FGF23 occurs first (for simplicity in this figure, decreased GFR has been selected as the initiating factor). The increase in plasma FGF23 concentration leads to decreased 1α-hydroxylase expression and therefore decreased plasma 1,25(OH)2D3 concentration, removing the negative feedback effect of 1,25(OH)2D3 on parathyroid hormone (PTH). In addition, plasma PTH concentration is increased due to decreased plasma calcium concentration, as a result of hyperphosphatemia and decreased plasma 1,25(OH)2D3 concentration. PTH results in an increase in the ratio of receptor activator of nuclear factor–κB ligand/osteoprotegerin (RANKL/OPG), which leads to increased osteoclastic bone resorption. The normal negative feedback effect of FGF23 on PTH production is lost, partially due to decreased expression of the FGF23-klotho-FGFR1c (fibroblast growth factor receptor 1c) in the parathyroid gland. A decrease in renal klotho also prevents the normal function of FGF23 to increase renal excretion of phosphorus. Chronic kidney disease causes an increase in wnt inhibitors, resulting in decreased wnt-β-catenin signaling and decreased bone formation. The combination of increased bone resorption and decreased bone formation results in the renal osteodystrophy seen microscopically. Increased plasma FGF23 concentrations are also a risk factor for left ventricular hypertrophy and vascular calcification in humans.
Bone turnover changes also occur prior to changes in plasma phosphorus, calcium, PTH, or 1,25(OH)2 D3 concentrations, possibly associated with changes in osteocyte biology. FGF23 and DMP1 expression is increased in osteocytes of trabecular bone in humans with early CKD-MBD. 144 Sclerostin is increased in early kidney disease, resulting in decreased signaling of the wnt/β-catenin pathway, increased receptor activator of nuclear factor (NF)–κB ligand (RANKL), and subsequently increased bone resorption, but the hypothesis is that coupling of bone resorption to bone formation means that bone formation is also temporarily increased, despite inhibition of the wnt/β-catenin pathway. 160 At this stage, perhaps increased sclerostin expression also contributes to the early increase in FGF23 expression. 157 As the kidney disease progresses, PTH increases, leading to increased bone resorption. PTH antagonizes sclerostin; therefore, decreases in sclerostin 14 should remove the inhibition of wnt signaling and bone formation. However, other wnt inhibitors, such as secreted frizzled related protein 4 (SFRP4), increase, so it is hypothesized that there is continued inhibition of wnt signaling, resulting in the decreased bone formation seen in patients (Fig. 5). 160
Traditionally, the decrease in serum 1,25(OH)2D3 concentration seen in chronic kidney disease was thought to be associated with decreased renal mass, but it is now far more likely that it is the result of inhibition of renal 1α-hydroxylase by increased FGF23. 76 Decreased 1,25(OH)2D3 leads to decreased feedback inhibition on PTH, resulting in increased PTH, increased osteoclastic bone resorption, and release of phosphorus, further increasing FGF23. 76,212 While normally FGF23 directly inhibits PTH production, the combination of decreased 1,25(OH)2D3 and decreased expression of the klotho/FGFR1c receptor complex in the parathyroid gland means that it has little effect on PTH secretion in advanced chronic kidney disease. 64,98 However, the recent discovery of a klotho-independent calcineurin-NFAT (nuclear factor of activated T cells) pathway for FGF23 in the parathyroid means that downregulation of the klotho/FGFR1c receptor complex does not completely explain the lack of response to FGF23 in the parathyroid gland in CKD-MBD, as this pathway is also involved in suppression of PTH secretion. 140 Renal klotho also decreases with increased stage of renal disease, possibly associated with decreased 1,25(OH)2D3; 61,124 this has consequences for the phosphaturic function of FGF23, further worsening phosphate retention and increasing plasma FGF23 concentrations. 163 In summary, FGF23 increases early in chronic kidney disease, possibly due to decreased renal excretion of phosphorus, resulting in decreased plasma 1,25(OH)2D3 concentration and increased PTH (Fig. 5).
Recent evidence also suggests that FGF23 is an independent risk factor for human cardiovascular disease. 90,204 Cardiovascular disease is common in chronic kidney disease, and increased serum FGF23 concentrations are associated with increased mortality due to cardiovascular disease. 73,91,142 Hyperphosphatemia is also a risk factor for cardiovascular disease and mortality in chronic kidney disease. 39,48,72,193 Excessive FGF23 results in left ventricular hypertrophy via a klotho-independent mechanism, likely via the calcineurin-NFAT pathway. 53,168 However, recent work has suggested that FGF23 also regulates the number of Na+-Cl– cotransporters in the distal tubules of the kidney. 4 In mice that overexpress FGF23, there was increased resorption of Na from the renal tubules, thus resulting in increased blood volume, hypertension, and ventricular hypertrophy, perhaps explaining part of the link between chronic kidney disease and increased cardiovascular risk. 4 Unfortunately, although administration of FGF23 antagonists decreases left ventricular hypertrophy and many of the features associated with secondary hyperparathyroidism in chronic kidney disease, mortality actually increases due to hyperphosphatemia and tissue mineralization. 168 Klotho may also have a part to play in the pathogenesis of cardiovascular disease in chronic kidney disease, as klotho inhibits vascular calcification 111 and is decreased in kidney disease; 80 however, conflicting results have been found in some studies, 112 and more work is required to unravel this mechanism.
Fibroblast Growth Factor 23 in Domestic Animals: What We Know to Date
While FGF23 is well characterized in mice, rats, and humans, 3,218,224 to our knowledge, the role of FGF23 in domestic animal physiology and disease has been little explored until recently.
FGF23 manipulation has been considered as a means of improving phosphorus utilization efficiency in nonruminant production animals (pigs and chickens), as phosphorus, due to cost and environmental concerns, is often a limiting factor in nonruminant diets 20,34 Chicks given an anti-FGF23 antibody and fed a phosphate-deficient diet had no differences in plasma phosphate concentration or bone ash percentage compared with chicks on a diet containing normal levels of phosphate. 20 Both acute and chronic phosphorus deficiencies occur in ruminants, but a recent review of phosphorus homeostasis and disorders in ruminants stated that the role of phosphatonins in ruminants had not yet been studied, 69 indicating a large deficit of knowledge in this area and a potential unexplored mechanism for the prevention of metabolic disease in early lactation.
As discussed earlier, inherited metabolic disorders such as ADHR cause a minority of human rickets or osteomalacia cases. At this time, comparable genetic disorders of FGF23 metabolism in animals have only been described in sheep. 43 Corriedale sheep with inherited rickets have a premature stop codon in the dentin matrix protein 1 gene, 230 equivalent to autosomal recessive hypophosphatemic rickets type 1 of humans. 55,118 While serum FGF23 concentrations have not been measured in affected sheep, it seems likely that the DMP1 mutation results in increased FGF23, leading to the phosphaturia, hypophosphatemia, and rickets seen clinically in affected sheep. 41 Most cases of inherited rickets described in domestic animals, however, involve defects in vitamin D metabolism or function, particularly mutations in the vitamin D receptor. 42
The existence of a phosphatonin system in dogs comparable to that in humans was suggested by an early study in which a 6-week-old puppy developed phosphaturia after being injected with an extract from a tumor that caused TIO in a human patient. 8 This result suggests that a phosphatonin bone-renal axis probably occurs in the dog.
Hemangiopericytomas and other soft tissue sarcomas, the traditional tumors associated with TIO in humans, are common in dogs. 44 Although TIO has not been reported in dogs or any other animal species, in our laboratory, we demonstrated gene expression of FGF23 by 15 of 49 canine soft tissue sarcomas (31%); 3 of these had high relative expression and some features resembling phosphatonin-expressing mesenchymal tumors of humans. 75 Clearly, further work is required to establish the importance of this finding; however, it may be that TIO is an unrecognized problem in a percentage of canine sarcoma patients, given the subtle radiographic signs of osteomalacia and common presentation of dogs for arthritic pain, which may easily be confused with bone pain. 188 However, TIO has never been diagnosed in dogs, and it should be considered that if FGF23-positive tumors are located in easily palpable subcutaneous sites, rapid recognition and surgical resection might allow insufficient time for osteomalacia to become clinically apparent.
Renal tubular phosphorus handling defects leading to hyperphosphatemia and periarticular calcification have been suspected in Hungarian Vizsla dogs, 50,143 but published evidence of an inherited phosphatonin disorder in this species is lacking.
FGF23 has been evaluated in cats with chronic kidney disease and/or hyperthyroidism. 40,58,66,67,209 FGF23 increases as the stage of kidney disease increases in cats. 66 No matter the stage of kidney disease, and despite similar plasma creatinine concentrations, cats with hyperphosphatemia had higher plasma FGF23 concentrations than did normophosphatemic cats. 66 Serum FGF23 concentrations also exponentially increased as glomerular filtration rate decreased, and this partially paralleled increases in serum creatinine concentrations. 58 Feeding a protein- and phosphate-restricted diet to cats with chronic kidney disease resulted in a decrease in plasma FGF23 concentration (regardless of the plasma phosphate concentration) and a decrease in plasma phosphate concentration (in those cats that were hyperphosphatemic) but no decrease in plasma creatinine concentration. 65 Plasma FGF23 concentrations may predict the future development of azotemia and chronic kidney disease in cats. In 1 study, cats that were initially nonazotemic but developed azotemia over the course of the following 12 months had higher plasma FGF23 concentrations at the beginning of the study than those cats that did not develop azotemia. 58
Similarly, hyperthyroid cats that developed azotemia and chronic kidney disease after treatment also had increased plasma FGF23 concentrations prior to instigation of treatment for the hyperthyroidism. 209 However, even hyperthyroid cats that did not develop azotemia had an increase in FGF23 concentrations—this was observed in the face of decreased phosphate and PTH concentrations. 209,210 This is not the case in humans with hyperthyroidism (Graves disease), 141,216 and the authors postulated some potential mechanisms to explain this, 209 such as the decline in glomerular filtration rate after treatment of hyperthyroidism, leading to a decreased filtering of FGF23, or possibly changes in 1,25(OH)2D3 concentration after treatment. However, a recent study investigating the relationship between 1,25(OH)2D3 and FGF23 in hyperthyroid cats showed there were no significant changes in either plasma 25OHD or 1,25(OH)2D3 concentrations pre- and posttreatment for hyperthyroidism. In addition, azotemia and plasma FGF23 concentrations showed no correlation with plasma 1,25(OH)2D3 concentration. 208
Conclusion
The discovery of multiple phosphatonin pathways has completely changed our understanding of phosphorus metabolism and opened up new avenues to explore with regard to treatment of chronic kidney disease and perhaps other diseases in which calcium and phosphate are deranged. Substantial work is required to determine if the phosphatonin pathways of veterinary species are similar to those in humans; certainly, we know that there are differences in vitamin D metabolism, 42 and so it stands to reason that given evolutionary adaptation, differences may also occur in the phosphatonin system.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.