
Abstract
The FAM20 family of secreted proteins consists of three members (FAM20A, FAM20B, and FAM20C) recently linked to developmental disorders suggesting roles for FAM20 proteins in modulating biomineralization processes. The authors report here findings in knockout mice having null mutations affecting each of the three FAM20 proteins. Both Fam20a and Fam20c null mice survived to adulthood and showed biomineralization defects. Fam20b –/– embryos showed severe stunting and increased mortality at E13.5, although early lethality precluded detailed investigations. Physiologic calcification or biomineralization of extracellular matrices is a normal process in the development and functioning of various tissues (eg, bones and teeth). The lesions that developed in teeth, bones, or blood vessels after functional deletion of either Fam20a or Fam20c support a significant role for their encoded proteins in modulating biomineralization processes. Severe amelogenesis imperfecta (AI) was present in both Fam20a and Fam20c null mice. In addition, Fam20a –/– mice developed disseminated calcifications of muscular arteries and intrapulmonary calcifications, similar to those of fetuin-A deficient mice, although they were normocalcemic and normophosphatemic, with normal dentin and bone. Fam20a gene expression was detected in ameloblasts, odontoblasts, and the parathyroid gland, with local and systemic effects suggesting both local and/or systemic effects for FAM20A. In contrast, Fam20c –/– mice lacked ectopic calcifications but were severely hypophosphatemic and developed notable lesions in both dentin and bone to accompany the AI. The bone and dentin lesions, plus the marked hypophosphatemia and elevated serum alkaline phosphatase and FGF23 levels, are indicative of autosomal recessive hypophosphatemic rickets/osteomalacia in Fam20c –/– mice.
Keywords
The FAM20 family of secreted proteins in mammals consists of three members (FAM20A, FAM20B, and FAM20C) that were initially thought to have roles in regulating the differentiation and function of hematopoietic and other tissues. 65 Since then, there have been reports linking mutations in both FAM20A and FAM20C to developmental disorders involving bones and/or teeth, suggesting a role for FAM20 proteins in modulating biomineralization processes. In humans, a mutation in FAM20A was recently linked to the occurrence of amelogenesis imperfecta and gingival hyperplasia in a consanguineous family. 69 However, lesions were not reported in bones or teeth of transgenic mice with a partial deletion of Fam20a, and their stunted growth was attributed to malnutrition and an unspecified metabolic disorder. 4 To the best of our knowledge, there have been only two reports elucidating the function FAM20B, which was shown to be a kinase that phosphorylates glycosaminoglycans 50 and in vivo to be involved in cartilage matrix production and skeletal development in zebrafish. 24 In contrast, there are several recent reports in the literature about FAM20C. The protein sequence of FAM20C is well conserved across species, with human and mouse sequences displaying 85% identity and 91% similarity. 86 The C-terminal half of the protein displays the highest level of conservation and this region has been termed the conserved C-terminal domain (CCD). 65 FAM20C (which was previously known as dentin matrix protein 4 [DMP4]) is a secreted protein expressed in osteoblasts, ameloblasts, and odontoblasts. 86 The localization of FAM20C in the matrices of dentin, enamel, and alveolar bone suggests that it is an extracellular matrix protein. 108 Mutations in FAM20C have been linked to Raine syndrome in humans, an osteosclerotic bone dysplasia, proving a role for FAM20C protein in bone development and mineralization. 36,86 Raine syndrome is accompanied by increased periosteal bone formation and facial dysmorphism. 86 Since first reported, 71 most described cases have been lethal during the neonatal period, characterized by dysmorphia, cerebral calcifications, choanal atresia or stenosis, and thoracic/pulmonary hypoplasia. Radiographic studies have shown a generalized increase in the density of all bones and a marked increase in the ossification of the skull. 1 Increased periosteal bone formation differentiates Raine syndrome from osteopetrosis and other known osteosclerotic bone dysplasias. 59 The periosteal bone formation typically extends along the diaphysis of long bones. 44 Affected individuals usually die at birth from respiratory failure, with autopsy findings of generalized osteosclerosis. 86 However, recent findings have shown a much wider clinical spectrum associated with FAM20C mutations in humans than the classic neonatal lethal presentation. 87 Nonlethal forms of Raine syndrome are characterized by mineralization defects, including generalized osteosclerosis, small teeth with enamel dysplasia, and ectopic calcifications of the intervertebral disks and kidneys. 30 Some cases have shown biochemical findings consistent with hypophosphatemic rickets. 87 So far, all identified Raine syndrome mutations involve the evolutionarily conserved amino acids located within the CCD of FAM20C. Since the original submission of this article, FAM20C has been identified as a kinase that phosphorylates proteins in the secretory pathways, including secretory calcium binding phosphoproteins (SCPP), that have a high affinity for calcium and regulate biomineralization. 100 Very recently, another line of FAM20C null mice were reported to develop hypophosphatemic rickets (but not osteosclerosis), along with a significant downregulation of osteoblast differentiation markers and a dramatic elevation of fibroblast growth factor 23 (FGF23) in the serum and bone. 109 However, dental lesions were not reported in these mice. 109
We report here a summary of findings in knockout mouse lines having null mutations affecting each of the three members of the FAM20 family of secreted proteins. Amelogenesis imperfecta (AI) was the major common phenotype in the two lines (Fam20a and Fam20c) that survived into adulthood. Mice provide particularly valuable models for investigating fundamental processes involved in normal and abnormal tooth development because the incisor teeth of rodents form and erupt continuously throughout life, permitting access to all stages of enamel and dentin production. The identification of mouse models displaying AI phenotypes may be expected to help identify the genes and developmental processes involved in the biomineralization of enamel and further elucidate the pathogenesis of the different types of AI. 12,33 The other pathological changes that we observed in developing teeth, bones, or blood vessels after functional deletion of Fam20a or Fam20c support a significant role for both of these genes in controlling biomineralization processes. Our findings illustrate the value of including pathology in high throughput phenotype screening protocols on genetically engineered mice in order to identify genetic factors involved in both normal and pathologic processes of mineralization.
Material and Methods
Mouse Production
The full allele designations for the three mouse knockout (HOM) lines described in this report are Fam20a tm1Lex, Fam20b Gt(GST_4283_D8)Lex1, and Fam20c tm1Lex, but for brevity we have used Fam20a, Fam20b, and Fam20c, respectively. The knockout allele for Fam20b was generated by gene trapping whereas the knockout alleles for both Fam20a and Fam20c were generated using homologous recombination (Fig. 1). Gene trapping was performed using strain 129S5SvEvBrd-derived embryonic stem (ES) cells obtained from the OmniBank library. 120 An intragenic gene trap mutation in Fam20b was selected from the OmniBank library by a BLAST search of all existing insertion site sequence tags using the Fam20b genomic interval as a query, which identified clone GST_4283_D8. The mutation in this clone was confirmed using inverse genomic polymerase chain reaction (PCR) as previously described. 35 Briefly, oligonucleotide primers complementary to the gene trap vector were used to amplify the vector insertion site from clone GST_4283_D8, which was then compared to mouse genome sequence assemblies to localize the insertion with respect to the exons and introns of the Fam20b gene.
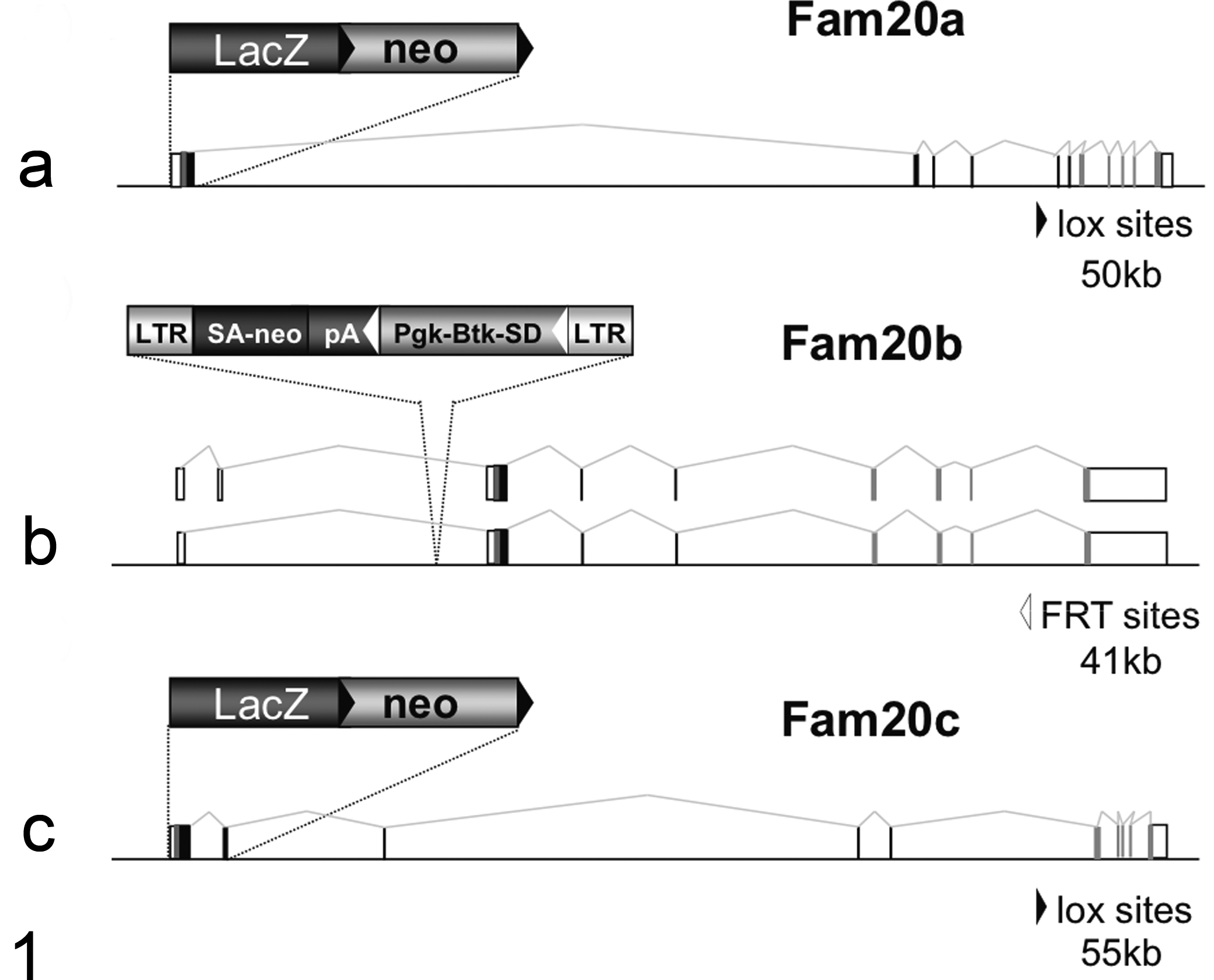
Generation of Fam20a –/–, Fam20b –/–, and Fam20c –/– mice. Overview of FAM20 gene mutations (a and c). Homologous recombination was used to replace the first coding exon(s) of Fam20a and Fam20c with a LacZ/neo selection cassette as shown. (b) Retroviral gene trap vector VICTR48 (EU676804) was used to produce OmniBank clone GST_4283_D8, which contains an insertion within intron 1 of the Fam20b gene, prior to the first coding exon (intron numbering based on accession NM_145413). Fam20b transcription/translation is disrupted through the action of the splice acceptor within the vector. Open boxes denote untranslated exons, filled boxes denote coding exons. The arrow denotes the location of the predicted signal sequence, the arrowhead denotes the conserved C-terminal domain. LacZ, beta-galactosidase gene; neo, neomycin phosphotransferase gene; LTR, viral long terminal repeat; SA, splice acceptor sequence; pA, polyadenylation sequence; Pgk, phosphoglycerate kinase-1 promoter; Btk-SD, Bruton’s tyrosine kinase splice donor sequence.
The first coding exon of Fam20a was deleted and replaced with an ES cell selection cassette containing the reporter gene LacZ and the neomycin gene using a pKO Scrambler NTKV-1901 vector (Stratagene, La Jolla, CA). In brief, genomic sequences upstream and downstream of the first coding exon of Fam20a were amplified by long-range PCR and were ligated into the pKO Scrambler vector along with the LacZ/neo ES cell selection cassette in a single unidirectional cloning step. For Fam20c, the first and second coding exons of gene were deleted. Briefly, a Lambda KOS phage library 110 was screened by duplex-PCR using oligonucleotide primers complementary to Fam20c. The genomic clone identified in this screen was cotransformed into yeast with a URA3 yeast selection cassette that had been appended with gene-specific arms flanking the first two coding exons. Clones that had undergone homologous recombination with the yeast selectable marker were isolated and the yeast cassette was replaced with an ES cell selection cassette containing the reporter gene LacZ and the neomycin gene. Completed pKOS or pKO Scrambler targeting vectors were electroporated into 129S5/SvEvBrd embryonic stem cells and G418/FIAU-resistant embryonic stem cell clones were isolated. Correctly targeted clones were identified and confirmed by Southern analysis (Fig. 2).
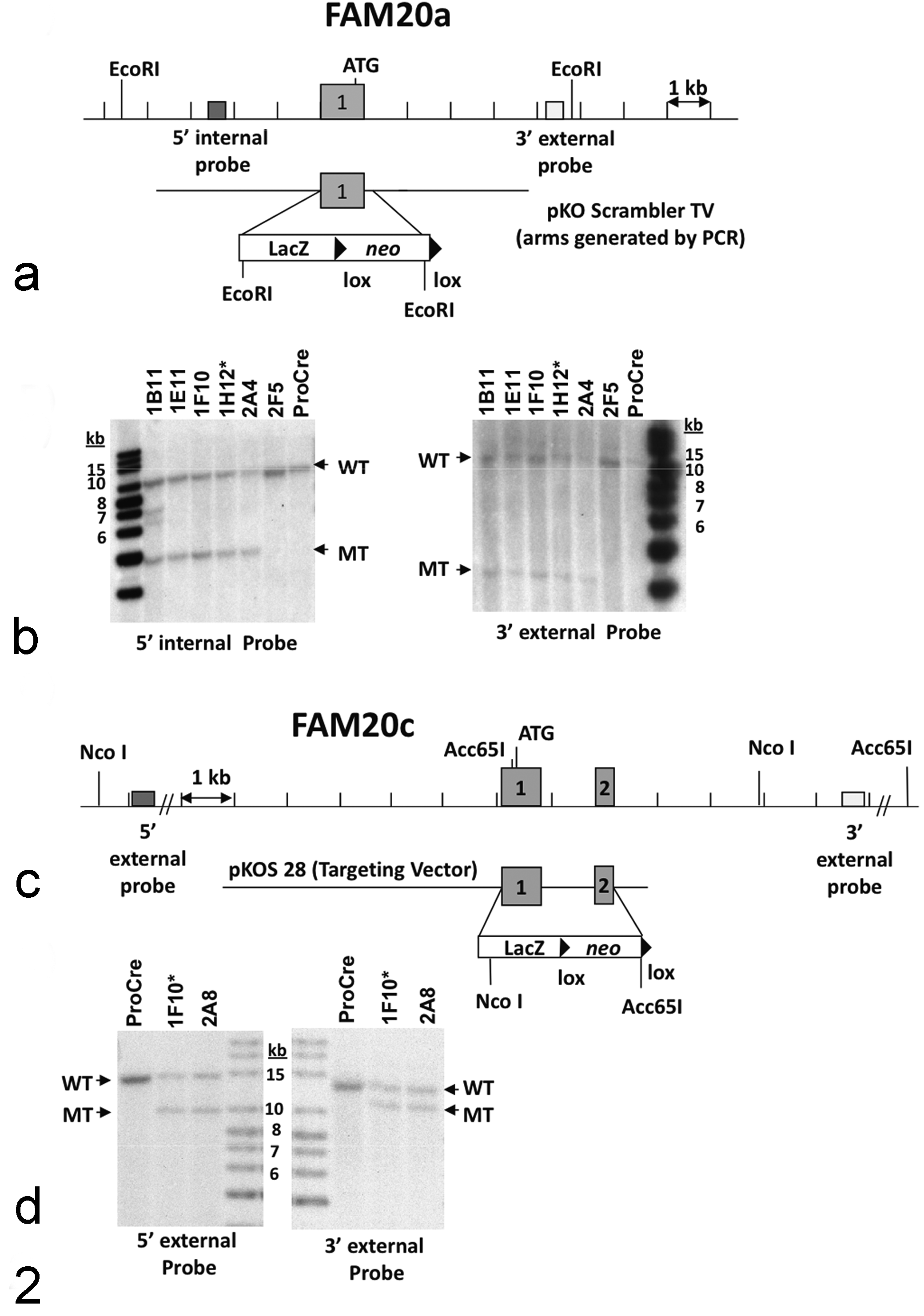
Targeting strategies and Southern blot confirmation of Fam20a and Fam20c gene mutations. (a) Homologous recombination between the pKO Scrambler targeting vector and the Fam20a gene resulted in the replacement of exon 1 with the selection cassette. Restriction enzyme recognition sites are indicated by EcoRI. ATG represents the translation start site. (b) Southern hybridization indicating Fam20a targeting in embryonic stem cell clones. Clones 1B11, 1E11, 1F10, 1H12, 2A4, and 2F5 were analyzed by Southern blot analysis using EcoRI digested genomic DNA with a 5′ internal probe and 3′ external probe. Three targeted clones were selected for blastocyst injections. An asterisk (*) indicates the clone that was transmitted through the germ line. (c) Homologous recombination between the pKOS targeting vector and the Fam20c gene resulted in the replacement of exons 1 and 2 with the selection cassette. Restriction enzyme recognition sites are indicated by Nco I and Acc65I. ATG represents the translation start site. (d) Southern hybridization indicating Fam20c targeting in embryonic stem cell clones. Clones 1F10 and 2A8 were analyzed by Southern blot analysis using NcoI digested genomic DNA with a 5′ external probe and Acc65 I digested genomic DNA with a 3′ external probe. The two targeted clones were used for blastocyst injections. An asterisk (*) indicates the clone that was transmitted through the germ line. WT indicates the band from the wild-type allele. MT indicates the band from the mutant allele. Band sizes are indicated in kilobases (kb) using a labeled size marker. ProCre represents untransfected embryonic stem cell DNA.
Targeted and gene trapped ES cell clones were microinjected into C57BL/6-Tyr c-Brd (albino) blastocysts to generate chimeric animals that were bred to C57BL/6-Tyr c-Brd (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous gene deficient mice. Knockout F2 mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (–/+) offspring of chimeric founder parents. Therefore, all mice used in these studies are of mixed C57BL and 129 genetic background. In all studies reported here, mutant mice were compared directly with their wild-type (WT) littermates used as negative controls. Using the albino variant of C57BL/6 mice (C57BL/6-Tyr c-Brd ) permits simple visual recognition of chimeric offspring because they have dark eyes and patches of dark hair that derive from stem cells from the agouti 129S5/SvEvBrd. Genotypes of offspring were determined by quantitative PCR as previously described. 35 Briefly, DNA isolated from tail biopsy samples was assayed by quantitative PCR for the neo gene, which is present in both the gene targeting and gene trapping vectors used to generate the mutations described in this study.
Mouse Husbandry and Testing
Mice were housed in micro-isolator cages within a barrier facility at 24ºC on a fixed 12-hour light and 12-hour dark cycle and were provided acidified water and Purina rodent chow No. 5001 (Purina, St. Louis, MO) ad libitum. All procedures involving use of live mice were conducted in conformance with Lexicon’s Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Quarterly sentinel surveillance showed no evidence of pathogenic rodent viruses, Mycoplasma, or Helicobacter species in the Lexicon Pharmaceutical source colonies. Lexicon Pharmaceutical’s phenotyping program included assays to evaluate a wide range of behavioral and physiological parameters intended to identify novel drug targets. In this program, screening assays were normally completed in mice ranging in age from 8 to 16 weeks, and knockout lines lacking potentially useful phenotypes were generally terminated after the initial screening process was completed. Clinical chemistry values, including serum alkaline phosphatase (ALP), calcium, and phosphorus, were measured using COBAS Integra 400 Analyzer (Roche Diagnostics, Indianapolis, IN). For serum phosphorus measurement in newborn mice, we used 1 microliter of serum in a sensitive malachite green colorimetric assay (Enzo Life Sciences, Farmingdale, NY). Serum FGF23 concentrations in young Fam20c –/– mice were measured using a commercial ELISA kit (Kainos Laboratories, Inc., Tokyo, Japan). MicroCT scans were obtained with a Scanco Medical µCT40 (Brüttisellen, Switzerland) with voxel dimensions of 6 micrometers. Dual-energy X-ray absorptiometry (DEXA) was performed on anesthetized mice (8 HOM and 8 WT) using a Lunar PIXImus instrument (GE-Lunar, Madison, WI). Statistical comparisons were made using the 2-tail Student’s t-test, based on inspection of group data and the assumption that the group data show a normal distribution.
Histopathology
Immediately after euthanasia, knockout mice and age-matched littermate control mice were routinely fixed by cardiac perfusion with 10% neutral buffered formalin. Tissues were collected and immersed in 10% neutral buffered formalin for an additional 48 hours except for eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, NY) overnight at room temperature. After overnight fixation, bones and heads were transferred into a decalcifying/fixation solution consisting of a mixture of formic acid and formaldehyde (Cal-Rite, Thermo Fisher Scientific, Waltham, MA), which allows simultaneous decalcification and fixation. In order to optimize the detailed evaluation of ectopic mineralization in soft tissues already embedded in paraffin blocks, surface decalcification was performed as follows: The block was sectioned to the desired levels and then placed face down in a small amount of decalcification solution (Cal-Rite) in a plastic Petri dish for 10 to 15 minutes. The block was then removed from the decalcification solution, rinsed well in water, and then placed on ice to cool before sectioning as usual. All tissues were embedded in paraffin, sectioned at 4 µm, mounted on positively charged glass slides (Superfrost Plus, Thermo Fisher Scientific, Waltham, MA), and stained with hematoxylin and eosin (H&E) for standard histopathologic examination. Special stains used included Masson’s Trichrome and Movat’s Pentachrome (both from Poly Scientific R&D Corp., Bay Shore, NY). For analysis of undecalcified bone, samples were embedded undecalcified in modified methyl methacrylate and sectioned with a Leica/Jung 2065 microtome. The bone sections were stained according to the Von Kossa method with a tetrachrome counterstain.
β-Galactosidase (LacZ) staining
β-Galactosidase reporter gene activity was assessed in situ as previously described. 105 Briefly, anesthetized mice were perfused sequentially with β-Gal fixative (0.2% glutaraldehyde, 1.5% paraformaldehyde, 2 mM MgCl2, 5 mM EGTA [ethylene glycol tetraacetic acid], 100 mM sodium phosphate [pH 7.3]); followed by 2 ml of β-Gal rinse (0.2% Nonidet-P40 (NP-40), 0.1% sodium deoxycholate, 2 mM MgCl2,100 mM sodium phosphate); and finally 10 ml of β-Gal stain (5 mM K3Fe(CN)6 [Potassium Ferricyanide], 5 mM K4Fe(CN)6 [Potassium Ferrocyanide], 1 mg of 5-bromo-4-chloro-3-indolyl-D galactopyranoside (X-Gal) (dissolved in dimethylformamide) per ml, 0.2% NP-40, 0.1% sodium deoxycholate, 2 mM MgCl2, 100 mM sodium phosphate [pH 7.3]). Tissues were post-fixed in β-Gal fix, rinsed in the β-Gal rinse, and then incubated in β-Gal stain solution for 48 hours. After 3 additional washes in β-Gal rinse, the tissues were post-fixed in Bouin’s fixative before dehydration and embedding in paraffin. Sections were cut at 4 µm and counterstained with Nuclear Fast Red (Vector Laboratories, Burlingame, CA).
Results
Fam20a–/– Mice
Although the number of heterozygous mice was significantly lower than expected (P = .0009, chi-square), Fam20a –/– mice were produced in approximately equal numbers to Fam20a +/+ mice (WT: 90; heterozygous [HET]: 119; HOM: 94) and displayed essentially normal growth rates and survival up to one year of age. No behavioral abnormalities were detected in any tests performed in Lexicon’s standard neurological phenotyping program. In addition, all immunological, physiological, metabolic, hematology, and blood chemistry parameters were within normal limits. Notably, blood levels of Ca, P, and alkaline phosphatase were normal (Table 1), and all measurements of bone parameters from microCT and DEXA were also within normal limits. Fertility in both males and females was also normal. In fact, the only notable findings identified during phenotyping of Fam20a –/– mice were discovered during histopathological evaluation. Initial pathological evaluations were completed on 12 Fam20a –/– mice (6 male and 6 female) aged between 14 and 20 weeks, while a second cohort of 12 Fam20a –/– mice (4 male and 8 female) was necropsied between 53 and 68 weeks of age.
Serum Ca, PO4, and Alkaline Phosphatase Levels in Fam20a Micea

Abbreviations: WT, wild-type; HET, heterozygous; HOM, knockout.
a All data shown represent means ± SEM for the number of samples indicated.
The most striking gross lesions were noted in the dentition of Fam20a –/– mice. Normal incisors in wild-type mice typically show yellow brown coloration and a smooth opalescent surface (Fig. 3). In contrast, both the upper and lower incisors in Fam20a –/– mice had a chalky white to opaque appearance with roughened/pitted surfaces from an early age (Fig. 4). The molar teeth of young Fam20a –/– mice were small and peg-like (Fig. 5), with an opaque and roughened surface and marked abrasion of the cusps, whereas the molar teeth of wild-type littermate mice were smooth and opalescent, with distinct cusps. In the older Fam20a –/– mice, the gross appearance of incisor teeth was essentially unchanged, but the molar teeth had worn down almost to the gum line.
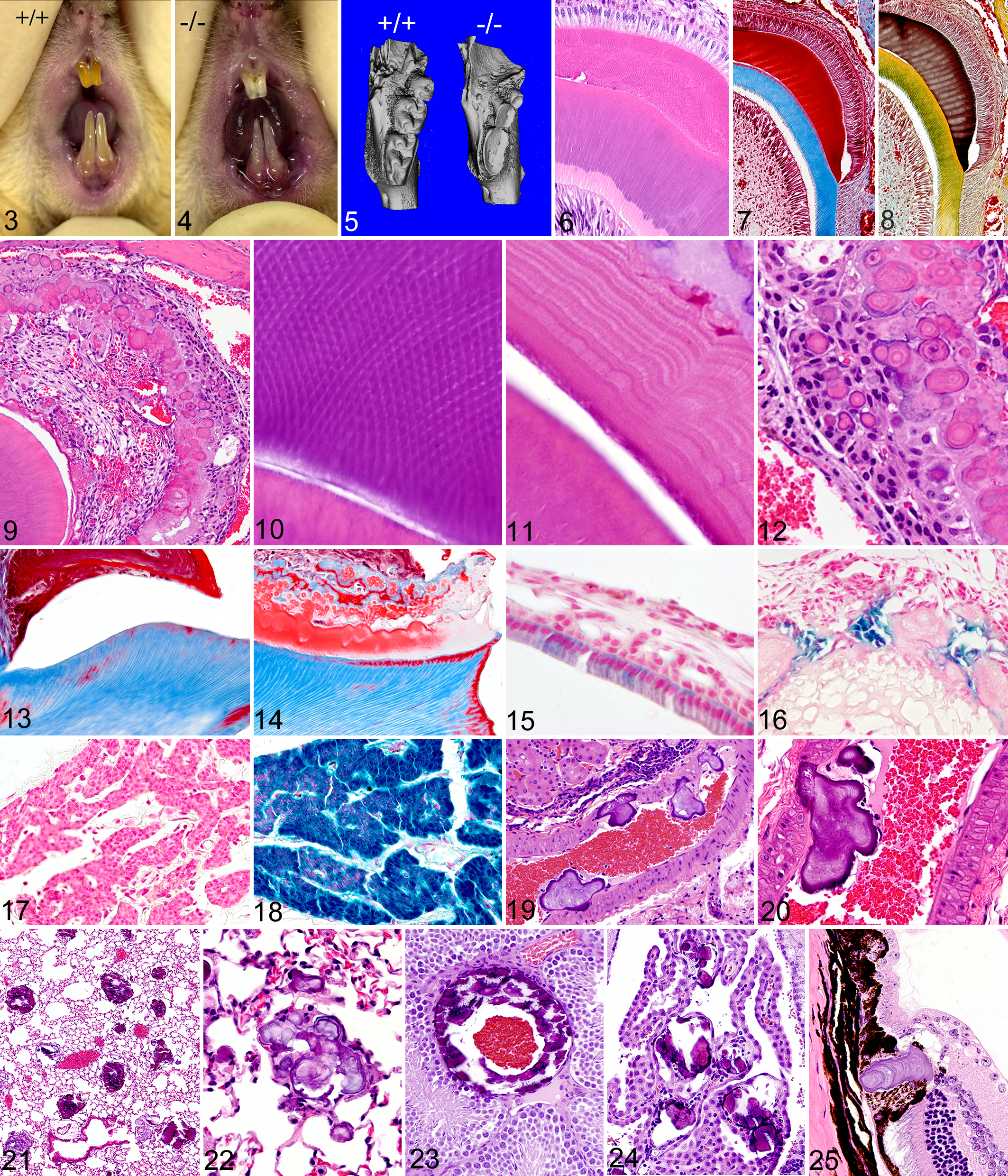
Incisor teeth; Fam20a+/+
mouse: The enamel of incisors in wild-type mice typically shows a smooth opalescent surface and has yellow brown coloration due to iron content.
Histologically, the normal developing incisor tooth consists of distinctive and easily identifiable layers of cells and extracellular matrix (Fig. 6). These layers can be highlighted with the use of special histochemical stains such as Masson’s Trichrome and Movat’s Pentachrome (Figs. 7, 8), and these special stains are useful for accentuating structural defects present in enamel and dentin. We found that tooth lesions in Fam20a –/– mice were essentially restricted to the enamel layer, with all incisor and molar teeth lacking true enamel. No abnormalities were detected in alveolar bone, cementum, dentin, or any other dental tissue aside from the enamel and enamel-forming layers.
Because incisor teeth in rodents grow continuously, it was possible to see the development of the enamel defect at different stages of tooth development within individual animals. In developing incisors, the ameloblast layer was markedly disrupted and disorganized (Fig. 9), and the decussating enamel rod substructure (Fig.10) often visible in normal developing teeth was absent. Instead, the irregular outermost layer of secretory ameloblasts and their associated enamel globules were often separated from the dentinoenamel junction by a central disorganized fibrovascular core. Enamel formation in Fam20a –/– mice appeared initially as irregular layers of eosinophilic enamel matrix oriented parallel to the dentinoenamel junction and lacking any discernable rod structure (Fig. 11). Ameloblasts in mutant mice initially appeared as palisades of tall columnar cells near the root of the incisor, but as enamel production progressed and became disrupted, the ameloblasts lost contact with the secreted enamel matrix at the dentinoenamel junction and became disorganized. The detached ameloblasts formed irregular nests of cells within a thin highly irregular mineralized crust that arose from the fusion of variably sized spherical to form poorly calcified multilobulated and lamellated enamel bodies (Fig. 12).
The various stages of enamel development in molar teeth were not assessed as they occur at fetal and neonatal stages. However, in decalcified sections of mature molar teeth of WT mice, the enamel layer appears as clear space, with the previous location and extent of the dissolved fully mineralized enamel often outlined by the remnants of surface enamel proteins and attached plaque (Fig. 13). In contrast, the decalcified molar teeth of Fam20a –/– mice still had irregular disorganized deposits of abnormal eosinophilic enamel matrix and lamellated globules covering most of the tooth surface and extending into the gingival sulcus and periodontal ligament (Fig. 14), indicating that enamel formation in Fam20a –/– molars was disrupted in a manner similar to that seen during incisor growth.
Fam20a gene promoter expression was analyzed in Fam20a –/+ and Fam20a –/– mice by using β-galactosidase enzyme histochemistry. Reporter gene expression was detected in both normal Fam20a –/+ (Fig. 15) and abnormal Fam20a –/– ameloblasts (Fig. 16). In the Fam20a –/+ mice, staining for Fam20a reporter gene expression was more intense in secretion-stage ameloblasts than in mature postsecretion-stage ameloblasts. Odontoblasts also consistently stained positively. Fam20a reporter gene expression was not detected by LacZ staining in any other tissues or cells in the body with one prominent exception; diffuse intense staining was present in the parathyroid gland in Fam20a –/+ and Fam20a –/– mice (Figs. 17, 18).
In contrast to the many abnormalities noted in the enamel of Fam20a –/– incisor and molar teeth, the structural arrangement and staining characteristics of dentin in the teeth of Fam20a –/– mice appeared to be indistinguishable from normal controls; in both wild-type and Fam20a –/– mice, primary dentin had a bone-like matrix arranged in columns between regular parallel dentinal tubules that traversed the entire thickness of the dentin layer. In Masson’s trichrome-stained sections, the primary and secondary tubular dentin underlying the tooth crown consistently stained bright blue. Given the absence of normal enamel in Fam20a –/– mice, secondary complications due to severe tooth wear were unexpectedly rare. Foreign material was not observed in the pulp cavity of any Fam20a –/– teeth, although mild periodontal inflammation surrounded a few molar teeth, pulp chamber necrosis and inflammation was found in only a single molar tooth in one of the older Fam20a –/– mice. Small pulp stones were present in a few molar teeth of the older mice but the dental pulp was otherwise normal.
The other major pathological finding in Fam20a –/– mice was the widespread and severe ectopic calcification of muscular arteries in most locations in the body, but most prominently in the kidneys. Grossly and histologically, the kidneys of approximately one-third of Fam20a –/– mice appeared essentially normal at gross examination, and the arcuate arteries in these kidneys contained only a few small calcified bodies. However, the other two-thirds of Fam20a –/– mice had small kidneys with pitted surfaces, which showed widespread calcification affecting virtually all small to medium size muscular arteries on histological examination. These vascular calcifications generally appeared as large irregular lamellated bodies that projected from the subendothelium into the vascular lumen (Fig. 19). In the smallest lesions, presumably at earliest stages of development, the calcifications were restricted to the intima and projected into the lumen. The central core of the lamellar bodies generally appeared to originate at the level of the intima or endothelium, with multiphasic addition of layers that resulted in large lamellated calcified bodies that projected into the lumen of affected blood vessels. In many locations, expanding calcifications were large enough to distort the shape of the affected artery and partially obstruct blood flow, but despite the large size and wide distribution of vascular calcification, areas of tissue damage and infarction were not seen outside of the kidney.
The protruding vascular calcifications were generally covered by endothelium, or sometimes partially surrounded by a thin layer of fibrin or cell debris. Although the subintimal and luminal vascular calcification often extended into and included the vascular smooth muscle (Fig. 20), dystrophic calcification of elastin in arteries was rarely seen, and the veins and elastic arteries were spared. The frequency, extent, and severity of vascular calcifications was greater in males than in females but did not appear to progress in severity in older mice. On the contrary, in the kidneys of the year-old mice, much of the vascular mineralization had resolved and had been replaced by irregular intramural foci characterized by chronic lymphoplasmacytic inflammation and fibrosis that extended from the vessel wall into the perivascular connective tissues. In the kidney, these chronic lesions were often associated with localized areas of parenchymal loss and collapse, with scattered dilated tubules containing eosinophilic proteinaceous fluid, suggesting multifocal infarction.
Ectopic mineralization was also a prominent feature in the lungs of Fam20a –/– mice, again being more common, severe, and widespread in male mice than in females. In females, the lamellated mineralized bodies in the lung were relatively small and rare (up to one per 20 40× fields). In contrast, pulmonary lamellar calcifications in male Fam20a –/– mice were larger and more widely distributed (Fig. 21), with at least a few being present in almost every 40× field. The smaller pulmonary calcifications initially appeared as lamellated round bodies confined within alveolar septa, but the larger bodies appear to have expanded into and merged with adjacent bodies to form large complex concretions that effaced and replaced alveoli in some areas (Fig. 22). Ectopic mineralization was also widespread in many other muscular arteries throughout the body. The arteries in the testes were particularly susceptible (Fig. 23), but calcification was found in the heart, skeletal muscle, subcutaneous adipose tissue, spleen, pancreas, thalamus, uterus, and choroid plexus (Fig. 24). In the eye, lamellated calcifications were consistently present within the ora ciliaris retinae (also known as the ora serrata), which is located at the junction of the neural retina and pars plana (Fig. 25).
The results of microcomputed tomography (μCT) scans supported and extended our histological findings. Hypomineralization of the defective enamel in all Fam20a –/– teeth was demonstrated both by the continued presence of eosinophilic matrix following decalcification (indicating high protein content) in histological sections and by its radiolucency in μCT scans (confirming low mineral content) (Fig. 26). No notable radiographic differences were observed in dentin or the surrounding alveolar bone in Fam20a –/– mice.
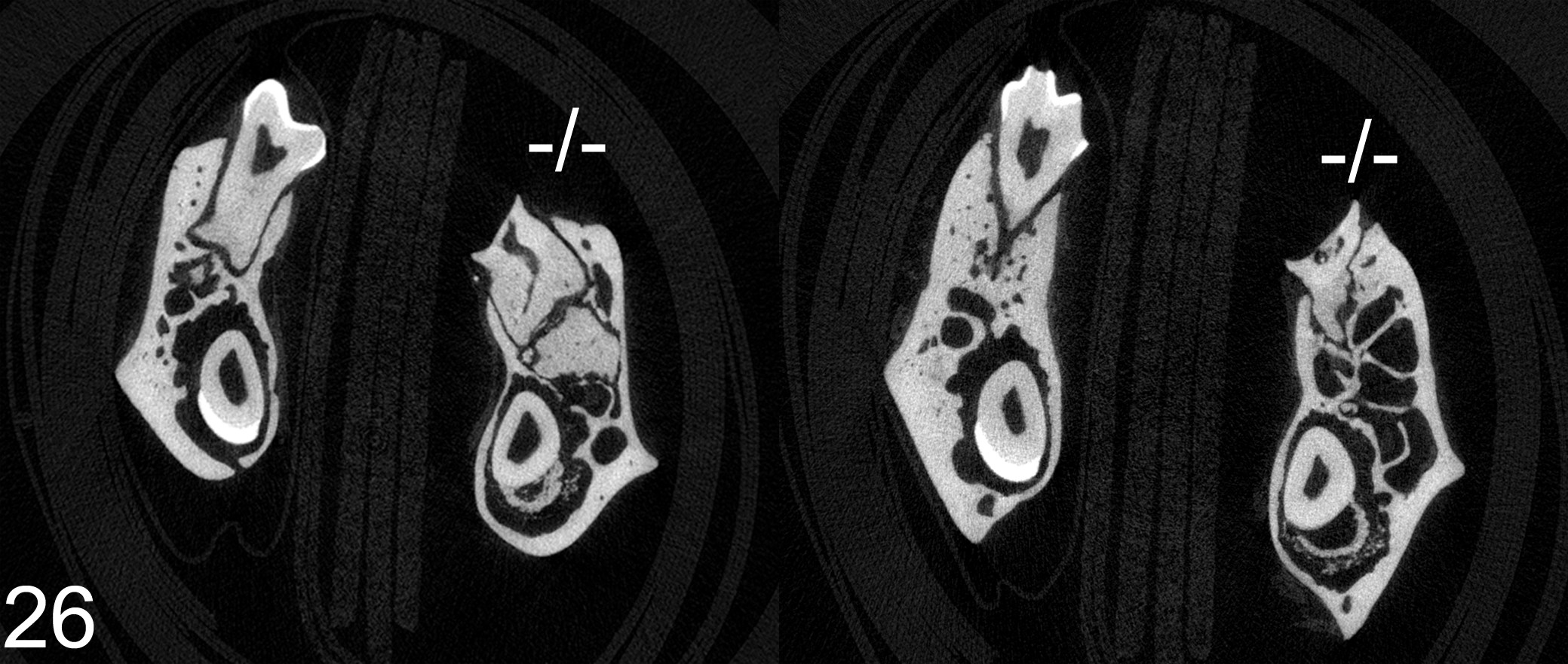
Mandibles with molar teeth; Fam20a+/+ and Fam20a–/– mice: μCT scans show the decreased radio-opacity of the abnormal enamel in both incisors and molars. No notable differences are present in surrounding bone or dentin of either wild type or knockout mice.
Fam20b–/– Mice
Few observations were made in the Fam20b –/– mice because deletion of this gene resulted in embryonic lethality. Genotypes were determined on embryos collected at 12.5 days post coitus; of these embryos there were 2 homozygotes, 20 heterozygotes, and 7 wild-types. There were also 7 resorption sites and one inconclusive placental site. The decreased viability, small size, and reduced numbers of mutant embryos suggest a critical role for this gene in embryological development. Histopathological evaluation of Fam20b –/– embryos showed severely stunted growth, with multisystem organ hypoplasia and delayed development most evident in the skeletal system, eyes, lung, GI tract, and liver.
Fam20c–/– Mice
In contrast to the largely normal gross appearance and behavior of Fam20a –/– mice, numerous phenotypic abnormalities were detected during regular phenotyping studies on Fam20c –/– mice. Prior to genotyping at 2 weeks of age, we observed an approximately 20% mortality rate in Fam20c –/– mice in more than 90 litters examined. The decreased viability of Fam20c –/– mice in the perinatal period was reflected in the abnormal Mendelian ratios observed (WT: 243; HET: 458; HOM: 191). Surviving Fam20c –/– mice had stunted growth, as shown by decreased mean body weight and body length at all time points. In addition, Fam20c –/– mice had bloated abdomens, developed balance problems, and often showed increased lacrimal discharge and crusting around the eyes. In neurological function testing, Fam20c –/– mice showed numerous abnormalities, including increased pre-shock, trace, and post-shock freezing during training in context trace conditioning and reduced mean ambulatory counts during one-hour habituation periods and during both light and dark phases. In open field testing, Fam20c –/– mice showed decreased median sum total distance. In pre-pulse inhibition testing, a lack of a startle response was suggestive of impaired hearing. Fertility of Fam20c –/– mice was not assessed due to early mortality.
DEXA scans showed decreased body mass measurements (including total tissue mass, lean body mass, total fat mass, percentage body fat) and reduced bone mineral content and density. MicroCT measurement in young male mice showed markedly decreased mean vertebral trabecular bone volume and connectivity density and reduced mean femoral midshaft cortical thickness. MicroCT analyses also revealed that bones were fragile, with many appearing to have been broken during dissection and collection. The microCT analyses showed a total or near total lack of dental enamel in the Fam20c –/– mice. Clinical pathology findings in adult mice showed markedly increased levels of alkaline phosphatase activity, along with mildly decreased Ca and markedly decreased P concentrations. Newborn Fam20c –/– mice were essentially normophosphatemic (WT = 6.1 ± 0.5, N = 5; HOM = 8.1 ± 1.5, N = 4; P = 0.21), but serum parameters measured at 18, 22, 26, and 30 days of age (Table 2) showed modest (11%) hypocalcemia, severe (55%) hypophosphatemia, and a 2.1-fold increase of serum alkaline phosphatase independent of age (P < 0.001) for all parameters (males and females combined). In addition, serum FGF23 levels measured at 30 days of age were 20-fold higher (9058 ± 3615 pg/ml, P < 0.01) in 6 hypophosphatemic Fam20c –/– mice than in 10 wild-type littermates (455 ± 88 pg/ml).
Serum Ca, PO4, and Alkaline Phosphatase Levels in Fam20c Micea
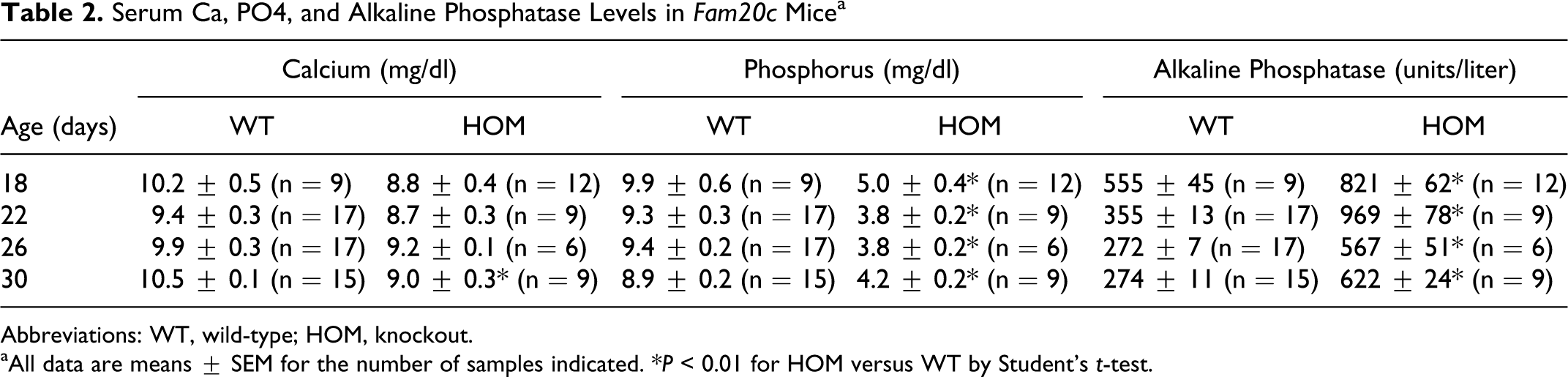
Abbreviations: WT, wild-type; HOM, knockout.
aAll data are means ± SEM for the number of samples indicated. *P < 0.01 for HOM versus WT by Student’s t-test.
Similar to the Fam20a –/– mice, the molar teeth of Fam20c –/– mice were small, peg-like, and had defective enamel. The poor condition of the teeth and severe endodontal and periodontal inflammation probably contributed to the reduced fat stores and poor growth of Fam20c –/– mice. Microscopic lesions in the enamel layer were essentially identical to those seen in the Fam20a –/– mice, but Fam20c –/– mice also showed pathological changes in the periodontal alveolar bone, dentin, and tooth pulp. The Fam20c –/– mice had abnormal enamel on all incisor and molar teeth. Normal developing incisor teeth have clearly defined layers consisting of an outer layer of tall columnar ameloblasts, followed by enamel, dentin, pre-dentin, and then odontoblasts (Fig. 27). In contrast, the ameloblast and enamel layers were severely disrupted in the developing incisor teeth of Fam20c –/– mice; changes in the enamel layer were characterized by the absence of an organized decussating enamel rod substructure and markedly disrupted ameloblastic epithelium (Fig. 28). Just as in Fam20a –/– mice, a thin band of eosinophilic enamel matrix was deposited in layers oriented parallel to the dentinoenamel junction in early stages of tooth development in Fam20c –/– mice. However, the secretory ameloblasts soon lost contact with newly secreted enamel matrix at the dentinoenamel junction and formed an irregular mineralized crust consisting of variably sized spherical and fused multilobular lamellated bodies similar to those found in Fam20a –/– mice (Fig. 12). Specialized histochemical stains showed that staining of normal secreted immature enamel (Fig. 29) was consistent with that of the lamellated globular bodies (Fig. 30, 31), which was crimson red with Masson’s trichrome stain and dark brown with Movat’s pentachrome stain. Hypomineralization of the defective enamel was evidenced histologically by its eosinophilia and continued presence on the surface of mature incisor and molar teeth following decalcification. Normal fully mineralized enamel appears histologically as a clear space in mature incisors and molars following decalcification. Enamel radiolucency in μCT scans proved its reduced mineral content in relation to dentin and bone. Unlike Fam20a –/– mice, the irregular cyst-like structures with fibrovascular cores were not present in the abnormal enamel layer of Fam20c –/– incisors.
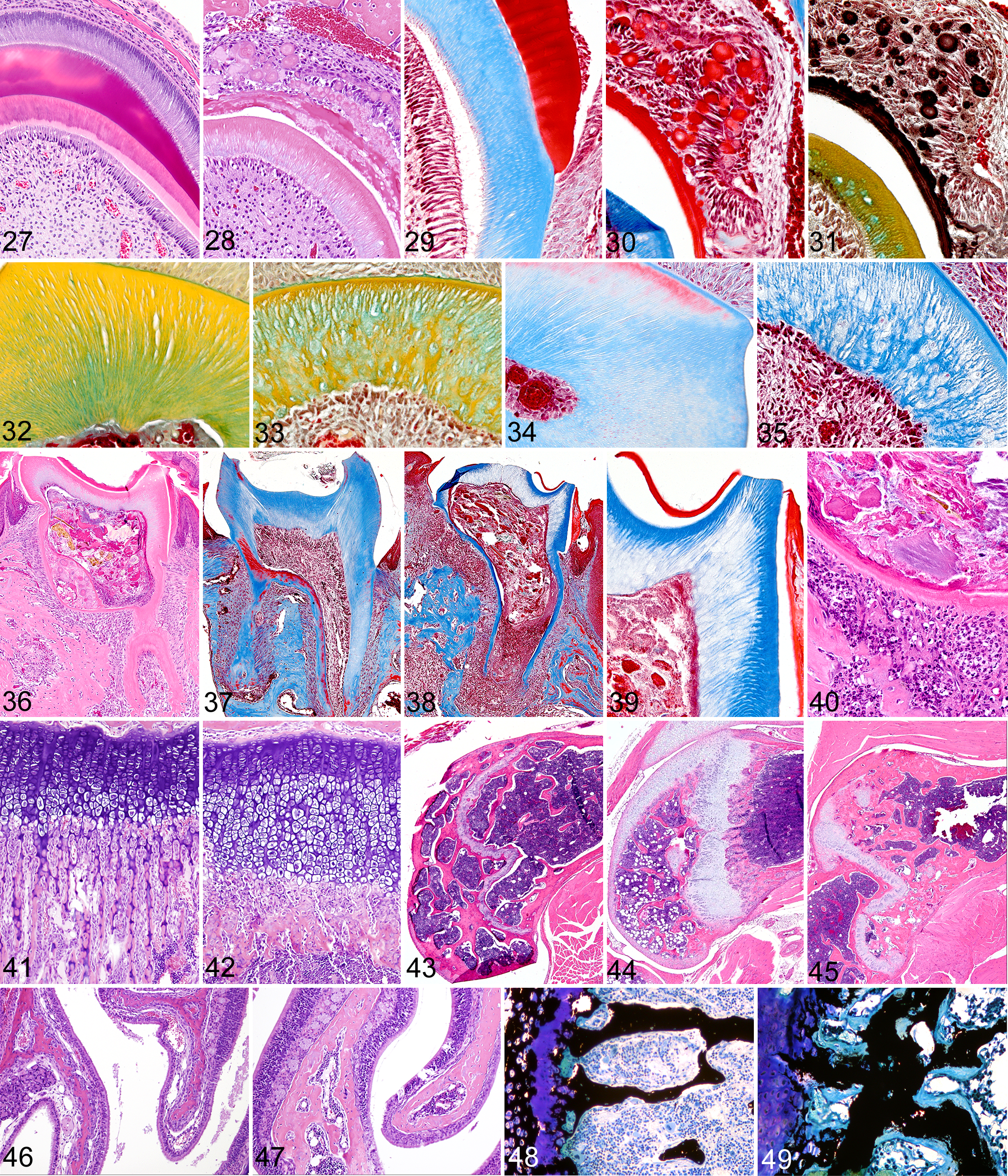
Incisor tooth; Fam20c+/+
mouse: Normal developing incisor teeth have clearly defined layers consisting of an outer layer of tall columnar ameloblasts, followed by enamel (red), dentin (pink), pre-dentin (light pink), odontoblasts, and dental pulp. H&E stain.
In addition to the defective enamel, numerous defects were evident in the primary and secondary dentin in both incisor and molar teeth of Fam20c –/– mice. However, since the original shapes and sizes of developing incisor and molar teeth were essentially normal, it appears that the original deposition of mantle dentin was unaffected. In incisor teeth, the defects in primary and secondary dentin initially appeared as scattered faintly basophilic vacuoles located within the matrix and wavy dentinal tubules in H&E stained sections. As the teeth matured, dentinal lesions progressed to loss of staining density and a decreased quantity and irregular distribution of dentin tubules. In both molar and incisor teeth, the irregular arrangements of dentin and the loss of staining density were highlighted effectively by both Movat’s pentachrome and Masson’s trichrome stains. With Movat’s stain, normal dentin has the appearance of regular parallel yellow columns (Fig. 32). In contrast, the dentin of Fam20c –/– mice appeared as irregular yellow swirls separated by loose pale green connective tissue (Fig. 33). With Masson’s Trichrome stain, normal dentin located below enamel caps is generally oriented as parallel blue columns (Fig. 34), whereas the abnormal dentin in Fam20c –/– mice appeared as irregular poorly stained strands separated by variably sized unstained areas (Fig. 35). The diffuse thinning of the dentin layer extended from crown to roots and resulted in enlarged pulp chambers, prominent pulp horns, and narrow irregular roots (Fig. 36). The roots of normal teeth often had areas of dentin that were thin and even stained red with Trichrome, but in all areas the dentin tubules extended from odontoblasts to the enamel or cementum (Fig. 37). Molar teeth of Fam20c –/– mice often had pulp necrosis associated with apical abscesses, periodontitis, and changes in alveolar bone (Fig. 38). The regular arrangement and relatively uniform staining of tubular dentin in normal molars was markedly different from the pale disrupted appearance of the thin dentin of Fam20c –/– mice (Fig. 39). In addition to highlighting the disorganization and reduction of primary and secondary tubular dentin, Masson’s trichrome staining also illustrated the absence of reactive or tertiary dentin in the pulp cavity in Fam20c –/– molar teeth and demonstrated clefts and fissures in many locations in the dentin. These dentin defects were often present underlying the cusps of molar teeth that permitted direct communication between the pulp and oral cavity, as shown by the presence of food particles and/or bacteria in many pulp cavities (Fig. 40). Periapical suppurative inflammation and foreign bodies often surrounded the molar tooth roots and inflammation extended into the periodontal alveolar bone (Fig. 40). Periosteal proliferation and lysis of tooth roots, cementum, and periodontal bone were associated with the periapical inflammation and necrosis, but other primary bone changes, characterized by markedly thickened maxillae and mandibles, were additional prominent features in Fam20c –/– mice.
Significant changes were noted in bones of both the young and mature Fam20c –/– mice. Examination of decalcified bone sections at 4 and 14 weeks of age showed that growth plate cartilage was thickened and disorganized, with marked changes being present in trabecular bone in both age groups. At the growth plate of long bones in wild-type 4-week old mice, the normal spongiosa consisted of long narrow trabecular extensions with delicate cartilaginous cores (Fig. 41). In contrast, Fam20c –/– mice had thickened irregular cartilaginous growth plates and relatively narrow spongiosa characterized by replacement of the normal arrangement of parallel thin primary trabeculae at the metaphysis by thick irregular transverse bands of osteoid forming subjacent to markedly thickened and irregular cartilaginous growth plates (Fig. 42). The normally narrow trabecular extensions of bone in the spongiosa were thickened and clubbed and there was a marked reduction of cartilage cores in newly formed trabeculae. There was also disorganization of capillaries, osteoblasts, and osteoid in the spongiosa, and although osteoclasts were present in large numbers, they appeared smaller than normal. Trabeculae were essentially absent in the diaphysis of long bones.
In the older mice, there was a progressive metaphyseal osteosclerosis. The narrow growth plate and fine trabeculae typical of normal mice (Fig. 43) transitioned from the thickened growth plate and short thick trabeculae of young mice (Fig. 44) to markedly widened trabeculae and an overall increased trabecular area in the metaphysis of long bones of mature mice (Fig. 45). In contrast, the trabeculae of flat bones (the sternum, ribs, calvarium) were notably thickened and extensive at both time points. The nasal turbinate bones of Fam20c –/– mice were also markedly thickened in comparison to those of wild-type mice (Figs. 46, 47). Examination of undecalcified sections of bone demonstrated that the thickened trabeculae contained extensive areas of unmineralized osteoid, consistent with hypophosphatemic rickets and osteomalacia (Figs. 48, 49). Even the ossicles in the middle ear were affected, with increased osteoid being present on the surface and at attachment sites. In contrast to the generalized thickening of trabecular bone, the cortical bone of the diaphysis of long bones was noticeably thinner than normal. The sternebrae were shorter and wider than normal, sometimes fused, and generally had increased osteoid mass due to wide trabeculae. In the older adult mice, osteoclasts were small, had scant cytoplasm, and appeared to be present in reduced numbers. Incidentally, the nasolacrimal ducts were dilated and filled with pale staining material in all of the older Fam20c –/– mice, suggesting the presence of distal duct stenosis, most likely due to thickening of the maxillary and turbinate bones.
In marked contrast to findings in Fam20a –/– mice, ectopic calcification was not a change observed in Fam20c –/– mice. In fact, the only focus of ectopic calcification in these mice was the delicate ring of lamellated mineralization present circumferentially in the eye at the junction of pars plana and retina (ora ciliaris retinae). Other notable pathology findings in Fam20c –/– mice included extramedullary hematopoiesis in the spleen and liver (secondary to periodontitis-driven granulocytopoiesis in the bone marrow) and severe hypoplasia of Leydig cells of the testes, seminal vesicles, and prostate gland, as well as the lack of secondary hormone-dependent differentiation of parietal epithelial cells of Bowman’s capsule in the kidney and granular cell tubular epithelium in the salivary gland of mature males. These findings suggest that testosterone production was markedly decreased from normal in mature Fam20c –/– mice.
Discussion
Physiologic calcification or biomineralization of extracellular matrices is a normal developmental process that is essential for the proper formation and functioning of various tissues (eg, bones and teeth). The pathological changes that develop in teeth, bones, or blood vessels after functional deletion of either Fam20a –/– and Fam20c –/– genes add support to previous reports indicating a significant role for their encoded proteins in modulating biomineralization processes. In the case of Fam20a –/– mice, the predominant lesions involved dental enamel and muscular arteries. However, Fam20a –/– mice were essentially normocalcemic and normophosphatemic, with normal dentin and bone. In contrast, Fam20c –/– mice were severely hypophosphatemic and developed notable lesions in both dentin and bone to accompany the enamel defects. Notably, the Fam20c –/– mice lacked foci of ectopic mineralization in the blood vessels and lungs. The embryonic lethality in our Fam20b –/– mice precluded detailed investigations, but the severe stunting in E13.5 Fam20b –/– embryos and the defects in cartilage matrix and perichondral bone formation reported recently in mutant zebrafish 24 support a possible role for this gene in skeletal development.
Bone, teeth, and otoconia are the only structures in the body that are normally mineralized, but any tissue can be abnormally mineralized as a consequence of genetic mutations involving a wide range of intercellular signaling and developmental gene networks. Although many of the specific lesions and target tissues affected by defective mineralization processes differed between Fam20a –/– and Fam20c –/– mice, the process of enamel formation was disrupted similarly in both knockout lines, resulting in AI. The process of amelogenesis is complex and thus is subject to disruption by several factors. Not surprisingly, AI in humans comprises a clinically and genetically heterogeneous group of disorders caused by mutations in genes that are critical for normal enamel formation and whose loss results in abnormalities in the amount, composition, and/or structure of enamel. Historically, use of the term amelogenesis imperfecta was restricted to hereditary disorders characterized by abnormalities in the quantity and quality of enamel unaccompanied by metabolic or morphological defects in other body systems. 116 However, the definition of AI has been expanded to include heritable enamel defects that may be associated with lesions affecting other organ systems. 2 To date, AI has been linked to mutations in six enamel proteins and proteinases (ENAM, AMBN, AMELX, MMP20, KLK4, and AMELOTIN), but many of the known AI kindreds do not have mutations in any of these genes. 16,77 Our findings in Fam20a –/– and Fam20c –/– mice strongly suggest that mutations in these genes might be responsible for some types of AI in humans.
The severe enamel defects and widespread mineralization of muscular arteries that we observed in Fam20a –/– mice suggest a local role for FAM20A in the tightly controlled and localized process of mineralization of enamel matrix as well as a systemic (possibly endocrine) role in preventing ectopic mineralization in the intima/subintima of blood vessels. The possibility of both local and systemic effects of FAM20A is supported by the distribution of β-gal reporter expression we observed, which showed Fam20a promoter expression to be restricted to ameloblasts, odontoblasts, and the parathyroid gland, with no detectable signal in endothelium or arterial walls. Interestingly, ectopic mineralization was not seen in Fam20c –/– mice, and Fam20c gene promoter expression was not found in the parathyroid gland. 108 However, Fam20c mRNA expression and FAM20C protein were detected in ameloblasts, odontoblasts, cementoblasts, and periodontal ligament fibroblasts, while extracellular FAM20C protein was localized within dentin, enamel, and alveolar bone, 108 consistent with the presence of dentin defects in Fam20c –/– mice. The recently reported function of FAM20C as kinase that phosphorylates secreted proteins that regulate mineralization 100 is consistent with our findings of local effects on enamel and dentin.
Both FAM20A and FAM20C are expressed by ameloblasts, and the development of AI in their absence shows that both are necessary for production of fully mineralized decussating enamel rods. In both cases, enamel rods are not formed and mineralization of the defective enamel matrix is abnormal. Elucidating the functions of FAM20A and FAM20C should enhance our understanding of the molecular mechanisms regulating enamel formation during tooth development. How disruption of these two genes causes AI might be better understood by comparisons with other defective enamel phenotypes associated with loss of specific ameloblast-derived proteins and briefly reviewing the processes of normal amelogenesis. Enamel differs from bone and dentin in its tissue origin, mineralization matrix, and mode of mineralization. Mineralization of bone and dentin proceeds within a collagenous matrix (osteoid and predentin, respectively) that is deposited by cells of mesenchymal origin and requires coordinated interactions between type I collagen matrix and several bone/dentin-specific noncollagenous proteins that help organize the matrix and regulate the mineralization process. 38 In bone and dentin, the major matrix constituent (collagen) remains embedded within the mineralized tissue. Type I collagen cannot induce matrix-specific precipitation of calcium phosphate on its own but relies on several different noncollagenous proteins that control nucleation and initial progression of mineralization. 31,32,45,107
In contrast, enamel mineralization takes place within a noncollagenous protein matrix that is secreted by ameloblasts of epithelial origin. This enamel matrix consists of specialized proteins that regulate the structural development and mineralization of enamel. 42 These protein components include enamelin (ENAM), 22 amelogenin (AMELX), 91 ameloblastin (AMBN), 51 amelotin (AMELOTIN), 47 and dentin sialophosphoprotein (DSPP). 16 Our findings indicate that FAM20A and FAM20C should be added to the list of proteins having a critical role in amelogenesis.
Newly secreted enamel matrix is composed largely of matrix proteins and has a soft gel-like consistency but also contains very small mineral crystals. 28 A distinguishing feature of enamel that differentiates it from bone and dentin is that extracellular matrix proteins are almost completely removed as the enamel protein gel matrix is converted into a structurally complex dense material consisting of tightly packed hydroxyapatite-like crystals. 7,88 Complete mineralization of the enamel matrix is delayed until the rods reach full thickness, and then the matrix is converted into hypermineralized inorganic enamel via a complex and tightly regulated process. Different components of the organic matrix of developing enamel control the initial nucleation and stabilize the growth of crystals. 23,76 Enamel rods that extend from the dentin layer to the surface of the tooth comprise the main structural feature of mature enamel, 19 with each rod containing tens of thousands of individual crystallites 18,66 of calcium hydroxyapatite (Ca10(PO4)6(OH)2), that together create the hardest biological tissue. 7,88
Deficiencies in any of the secreted ameloblast proteins result in the production of defective enamel. In recent years, mice deficient for amelogenin, 34 kallikrein 4 (KLK4), 84 matrix metalloproteinase 20 (MMP-20), 12 ameloblastin, 111 and enamelin 43 have been shown to develop AI phenotypes. The dental lesions displayed by each of these models have helped elucidate the functions of these matrix components in enamel formation. For example, amelogenin-deficient mice have very thin and rodless enamel. 34 The two secreted proteinases KLK4 and MMP20 enable the maturation (expansion) of enamel crystallites by removing matrix proteins. 56 Of these, MMP-20 is an early protease secreted along with enamel proteins by secretory-stage ameloblasts, 89 whereas the protease KLK4 is secreted later by transition- and maturation-stage ameloblasts and degrades the remaining organic matrix proteins following the termination of appositional growth of enamel rods. 89 The absence of KLK4 results in frail, hypomineralized enamel with normal rod/inter-rod organization and thickness. 84 MMP-20 deficiency produces thin enamel with disrupted rod patterns, 12 while deficiencies in either ameloblastin or enamelin result in formation of an abnormally thin, disorganized, and poorly mineralized enamel layer. 43,111 The disorganized thin layer of calcified material covering the incisor and molar teeth of both ameloblastin and enamelin deficient mice only contains 68% mineral by weight (similar to bone). 90 The precise mechanisms are not yet understood, but it appears that ameloblastin and enamelin must be present within the enamel matrix for the mineral-induction event to occur normally. 90 Defective enamel formation in ameloblastin/enamelin-deficient mice might result from reduced numbers of mineral crystallites per unit volume of enamel at the mineralization front. 90
Abnormalities at any stage of amelogenesis may result in AI, and most classification schemes to identify distinct subtypes of AI are based on clinical phenotype and mode of inheritance. 68,116 Generally, most morphological enamel defects can be characterized as having some combination of hypoplastic, hypomaturation, and hypomineralized enamel phenotypes. In the hypoplastic type, defective enamel matrix synthesis results in thin or weak enamel due to reduced apposition of enamel during tooth maturation. Hypoplastic enamel is thin, but its radiodensity remains greater than that of the underlying dentin. Hypomature enamel can be of normal thickness, but it is soft because the incomplete removal of proteins during the maturation phase results in incomplete and defective development of enamel crystals. The radiodensity of hypomature enamel is reduced to approximately the same as that of the dentin. Due to defective mineralization of the matrix and crystals, hypomineralized enamel has lower radiodensity than dentin and is soft, rough, and rapidly lost by attrition. Hypoplastic (thin but seemingly correctly mineralized) or hypomineralized enamel are the most common types of AI seen in humans, but an overlap of these characteristics is often present. 5
The abnormal enamel layer in Fam20a –/– and Fam20c –/– mice appears to form a weak, poorly mineralized crust that crumbles and is lost during mastication. In both lines, there is disruption of the mineralization front and rod formation at the secretory surface of the ameloblast, where the organization and elongation of enamel into rods is established. Taken together, our findings suggest that both Fam20a –/– and Fam20c –/– mice develop a phenotype characterized by a combination of enamel hypoplasia and hypomineralization, which is most similar to ameloblastin/enamelin-deficient mice.
In addition to AI, Fam20a –/– mice had striking lesions of ectopic mineralization, characterized by large intravascular calcifications of muscular arteries located within many tissues and scattered intrapulmonary calcifications in the lung. The widespread distribution of ectopic calcifications in Fam20a –/– mice in spite of essentially normal levels of calcium ion (Ca+2) and inorganic phosphate (Pi) in the blood coupled with the observation that FAM20A expression appears to be restricted to ameloblasts/odontoblasts of teeth and the parathyroid gland suggests that FAM20A has both local and systemic effects in controlling biomineralization.
For many years, the development of vascular and soft tissue calcifications was thought to be a passive process caused by the uncontrolled precipitation of calcium phosphate crystals when calcium ion (Ca2+) and inorganic phosphate ion (abbreviated as Pi) concentrations became elevated under certain pathological conditions. However, given that physiological concentrations of Ca2+ and Pi in serum are already high enough to result in spontaneous precipitation of calcium phosphate, it became clear that biomineralization must be tightly controlled by other local and systemic factors that either promote or inhibit mineralization. It was discovered that ectopic calcification could be triggered by the loss of powerful inhibitors that play critical roles in preventing calcification of soft tissues under physiological conditions, and that some of these same inhibitors also influence biomineralization processes in bones and teeth. Mouse models have helped elucidate the genetic and molecular basis of vascular calcification, and our findings indicate that FAM20A should be added to the list of known inhibitors of ectopic calcification, which presently includes inorganic pyrophosphate (the anion P2O7 4−, abbreviated as PPi), matrix-Gla protein (MGP), osteoprotegerin (OPG), osteopontin (OPN), and fetuin. We believe that FAM20A might interact with one or more of these calcification inhibitors, whose functions and relevant phenotypic effects in murine models will be described in more detail in the following.
While Pi is a critical promoter of mineralization, inorganic pyrophosphate (P2O7 4−, abbreviated as PPi) is probably the most important inhibitor of ectopic mineralization. PPi prevents mineralization by inhibiting initiation and/or propagation of hydroxyapatite crystal formation 64 and has been shown to prevent precipitation of saturated solutions of Ca and PO4 in plasma 61 and blood vessels. 101 Increasing levels of PPi also stimulate production of osteopontin, which is another inhibitor of nucleation. 92,106 Three different enzymes have been found to modulate PPi and Pi levels in mineralizing tissues such as bones and teeth. First, extracellular PPi is synthesized by the enzymatic hydrolysis of ATP by ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1, also known as NPP-1). 40 Second, secretion of intracellular PPi is controlled by the transmembrane-spanning cell surface protein ANKH, which is the human homolog of the mouse progressive ankylosis protein (ANK). 41 Lastly, the breakdown of extracellular PPi to phosphate is mediated by alkaline phosphatase (ALP), also known as “tissue nonspecific” alkaline phosphatase (TNAP), which hydrolyzes PPi to Pi, which then combines with calcium to generate hydroxyapatite. 115 Therefore, increased activity of both ENPP1 and ANK prevents ectopic calcification, whereas increased TNAP activity favors calcification by removing PPi, which is a potent inhibitor of calcification. 98 Recent findings indicate that vascular calcification is inhibited by the local synthesis and hydrolysis of PPi by the smooth muscle cell ENPP1 and TNAP, respectively. 104
Conversely, ectopic calcification of tissues is promoted by deficiencies in either ENPP1 37,48,75 or ANK. 41 In humans, idiopathic infantile arterial calcification has been linked to low levels of PPi resulting from deficient ENPP1 activity, 75 and certain loss-of-function mutations in ENPP1 have been found in hereditary hypophosphatemic rickets. 55 Recently, hypophosphatemia and altered bone development Enpp1–/– mice was linked to an increase in fibroblastic growth factor 23 (FGF-23), 58 which will be discussed in more detail in the following. The high prevalence of vascular calcification in chronic kidney disease patients on dialysis is associated with reduced plasma PPi levels, 53 and recent findings in mice suggest that vascular calcification might be prevented by inclusion of PPi in dialysis solutions. 73 In mice lacking functional ANK, there is intracellular trapping of PPi and reduced levels of extracellular PPi, which leads to ectopic mineralization in many locations. 41 Interestingly, vascular mineralization does not occur in Ank–/– mice, suggesting that there is sufficient ENPP1-mediated generation of PPi to prevent ectopic calcification in vessel walls. Recently, mutations in ANKH were linked to an autosomal recessive disease in humans characterized by mental retardation, deafness, ankylosis, and dental abnormalities, 62 while mutations in TNAP have been linked to hypophosphatasia, increased PPi, and osteomalacia. 113
MGP is a second potent inhibitor of mineralization in arteries and cartilage. 57 In MGP-deficient mice, vascular calcification starts in the arterial media and spreads rapidly along elastin fibers. 57 MGP-deficient mice also develop widespread mineralization of cartilage in the trachea and ears and excessive mineralization of the growth plate cartilage, which reduces longitudinal growth and results in osteopenia. The mineralization of elastic lamellae in arteries is often accompanied by chondrocytic transformation of vascular smooth muscle cells. It is known that MGP normally inhibits the differentiation of osteogenic and chondrogenic cells by sequestering BMP-2. 10 We found that Fam20a –/– mice do not have mineralization of elastic lamellae or chondrocytic differentiation of vascular smooth muscle, suggesting that FAM20A acts by a different mechanism than MGP. The potential role of MGP in vascular calcification in humans remains unclear because individuals lacking functional MGP do not have vascular calcifications, although they do develop ectopic calcification of cartilage. 63
OPG is a circulating glycoprotein that acts as a decoy receptor for RANKL and prevents the differentiation and activation of bone-resorbing osteoclasts by blocking the binding of RANKL to its receptor RANK. The direct effects of OPG on osteoclastogenesis are demonstrated by the development of osteoporosis in OPG-deficient mice and conversely, by the development of osteopetrosis in transgenic mice that overexpress OPG. 85 In addition to its important role in osteoclastogenesis, OPG is involved in vascular calcification, 82 and OPG-deficient mice develop both osteoporosis and medial calcification of the aorta and renal arteries. 11 Compared to MGP-deficient mice, the vascular calcification due to OPG deficiency is rather mild and restricted in extent; nevertheless, it suggests that OPG or its ligands play a role in the association between osteoporosis and vascular calcification. 11 However, the mechanisms by which OPG induces vascular calcification are still unclear, and its multiple roles in mediating vascular pathology are still under investigation. 17 We feel that it is unlikely that FAM20A and OPG have a direct relationship because of significant phenotypic differences in mice deficient in these proteins. The vascular mineralization in Fam20a –/– mice affects muscular arteries in all parts of the body and is not associated with either aortic lesions or osteoporosis. In addition, the distinctive pulmonary calcifications seen in Fam20a –/– mice have not been reported in OPG deficient mice.
OPN is a secreted acidic phosphoprotein and a component of bone, where it facilitates the attachment of osteoclasts to the bone matrix. 13 OPN is a potent inducible inhibitor of vascular calcification, apparently by binding to and physically blocking hydroxyapatite crystal growth, as well as by inducing the expression of carbonic anhydrase II in monocytic cells and promoting acidification of the extracellular milieu. 94 Carbonic anhydrase–deficient mice show age-dependent medial calcification in arterioles and small arteries (including renal hilar and arcuate arteries) and osteopetrosis. 93 In OPN/MGP double null mice, mineralization of vessels was more rapid in onset and severe than in mice deficient in MGP alone. 92 Nevertheless, OPN-deficient mice have relatively subtle mineralization abnormalities that are much milder than what we observed in Fam20a –/– mice. 37
The final inhibitor of calcification to be discussed is fetuin-A, which is a circulating glycoprotein synthesized in the liver, secreted into blood, and, having a high affinity for hydroxyapatite, is incorporated into bone during mineralization. 102 Fetuin-A prevents ectopic vascular calcification by inhibiting the precipitation of hydroxyapatite crystals in supersaturated solutions of Ca and Pi ions. 39,80 This inhibition of calcification is mediated by calciprotein particles, which are composed of fetuin-A and colloidal basic calcium phosphate. 39 Fetuin A is also involved in the regulation of osteogenesis and bone resorption. 99 Clinical studies consistently show that fetuin-A deficiency is associated with vascular calcification and mortality in chronic kidney disease patients. Both chronic inflammation and uremia may contribute to fetuin-A depletion, 98 and low fetuin-A serum concentrations in hemodialysis patients are associated with severe extraosseous calcifications and highly increased cardiovascular mortality. 49
Fetuin-A’s function as a potent inhibitor of mineralization has been demonstrated in mice. On standard diets, fetuin-A–deficient mice on most genetic backgrounds display a mild phenotype characterized by growth plate defects, increased bone formation with age, and enhanced cytokine-dependent osteogenesis. 99 However, fetuin-A–deficient DBA/2 mice develop widespread dystrophic soft tissue calcification in the kidney, lung, and heart. 78 The spontaneous development of ectopic calcifications in fetuin-A–deficient mice is dependent of a DBA/2 genetic background, which makes them naturally predisposed to pathological calcifications. 78 In contrast, fetuin-A–deficient mice on a C57Bl /6 genetic background are resistant to ectopic calcification, which could be induced only by feeding them a diet containing elevated levels of phosphate and vitamin D. 78
The location and types of ectopic mineralization that we observed in Fam20a –/– mice were most similar to those reported in fetuin-A–deficient mouse models, which showed calcification of muscular arteries and virtually identical pulmonary calcifications. Importantly, we observed spontaneous ectopic mineralization in Fam20a –/– mice bred on a hybrid but “calcification-resistant” C57Bl/6/129 genetic background and fed standard rodent chow. The phenotypic similarities between our Fam20a –/– mice and fetuin-A knockout mice suggest a possible close interaction between these two proteins and lead us to speculate that FAM20A secreted by the parathyroid gland may modulate the production, post-translational modification (phosphorylation), and/or release of fetuin-A. Future studies to measure fetuin-A responses to administration of FAM20A protein will be needed to confirm this hypothesis, but finding very similar pathology phenotypes in two different lines of knockout mice commonly indicates that they have homologous functions, are in the same gene family, or are active in the same metabolic pathway. 81 The partial resolution of ectopic mineralization in the muscular arteries and lungs as the mice matured indicates that age-related factors must also be involved in the pathogenesis of these lesions in Fam20a –/– mice. We also observed that male Fam20a –/– mice are more susceptible than females to ectopic mineralization. It is possible that higher levels of other calcification inhibitors in female mice could be responsible for this protective effect. For example, in humans, serum concentrations of fetuin-B (a less active inhibitor of calcium phosphate precipitation than fetuin-A) are higher in females than in males. 21
Although both Fam20a –/– and Fam20c –/– mice had virtually identical enamel defects, only the Fam20c –/– teeth consistently developed endodontitis and necrosis associated with foreign body material in the pulp cavity. All of the Fam20c –/– mice examined in this study developed primary dental lesions and associated suppurative periodontitis and thickened periodontal alveolar bone. In contrast, only a few teeth in all the Fam20a –/– mice showed endodontal lesions or periodontal inflammation despite having equally severe enamel defects. These markedly different outcomes in two different knockout mouse lines showing equally severe AI are most likely due to concurrent structural defects in the dentin of Fam20c –/– mice that were absent in Fam20a –/– mice. The dentin lesions in Fam20c –/– mice were evident at the earliest stages of development in both incisor and molar teeth. These defects in primary and secondary tubular dentin, as well as the absence of reactive tertiary dentin in the pulp cavity, indicate that Fam20c –/– mice have a primary defect in dentin development. The clefts and fissures within the dentin underlying the cusps of molar teeth appear to provide a direct route between the pulp and oral cavity, which permitted the entry of food particles into most pulp cavities, resulting in inflammation and necrosis. The periosteal proliferation and lysis of tooth roots, cementum, and periodontal bone were mostly due the periapical inflammation and necrosis extending from the pulp cavity, although a role for primary defects in these locations cannot be ruled out given the reported expression of FAM20C in ameloblasts, odontoblasts, cementoblasts, and periodontal ligament fibroblasts. 108 The markedly thickened maxillae and mandibles were a prominent feature in Fam20c –/– mice and mirrored similar changes in other bones throughout the body, where trabeculae were outlined by broad bands of osteoid.
The concurrent appearance of primary pathological defects in both dentin and bone in Fam20c –/– mice is not surprising, as genetic disorders affecting biomineralization often produce lesions in both dentin and bone. 70 FAM20C mutations in humans have been linked to Raine syndrome, which is characterized by osteosclerotic bone dysplasia and other deformities that are usually lethal in infants. However, recent findings indicate that not all FAM20C mutations result in death in infancy and that the broader range of possible phenotypes may include hypophosphatemic rickets.30,87 In young Fam20c –/– mice, the pathological changes in bones were typical of those associated with hypophosphatemic rickets. Long bones showed thickened and irregular growth cartilage, with shortened and fused trabeculae lacking distinct cartilage cores at the growth plate. With increasing age there was a progressive enlargement or flaring of the metaphysis, which was filled with broad bands of osteoid (osteomalacia). Although irregular trabecular bone/osteoid in the medullary regions of bone was often markedly thickened, the diaphyseal cortical bone was generally thinner than normal. These changes in bones of Fam20c –/– mice, plus the marked hypophosphatemia and elevated serum alkaline phosphatase and FGF23 concentrations, were changes consistent with a diagnosis of autosomal recessive hypophosphatemic rickets/osteomalacia.
The analysis of genetic disorders resulting in hypophosphatemic rickets has led to the identification of several proteins that are essential in regulating phosphate homeostasis. 97 Circulating factors that promote phosphate excretion and impair bone mineralization have been termed phosphatonins. 117 Hormones that specifically regulate Pi homeostasis (“phosphatonins”) currently include fibroblast growth factor 23 (FGF23), and matrix extracellular phosphoglycoprotein (MEPE), as well molecules that affect FGF23 and MEPE function, such as phosphate-regulating gene with homologies to PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) 14,15 and dentin matrix protein 1 (DMP1). 29 The principal phosphatonin is FGF23, which inhibits phosphate reabsorption by the sodium/phosphate cotransporter in the kidney, by acting through specific FGF receptors with the co-factor klotho protein. 72 FGF23 is made by osteocytes and activates KLOTHO/FGFR1 receptor heterodimers in the kidney to inhibit phosphate reabsorption and 1,25-dihydroxyvitamin D synthesis in the proximal renal tubules. 3,97
Study of the hereditary hypophosphatemic disorders in humans, and their corresponding mouse models, has contributed immeasurably to our understanding of several fundamental physiological processes involved in phosphate homeostasis and its influence on biomineralization. X-linked hypophosphatemia (XLH), autosomal recessive hypophosphatemic rickets (ARHR), and autosomal dominant hypophosphatemic rickets (ADHR) are the most common hereditary hypophosphatemic disorders. XLH is caused by mutations of PHEX 14,15 that affect the degradation and production of FGF23, causing increased circulating levels of the phosphatonin FGF23. 9 ARHR is caused by inactivating mutations to dentin matrix protein 1 (DMP1). 27,54 DMP1 is produced by osteoblasts/osteocytes and accumulates in the extracellular matrix, where it facilitates mineralization of collagen in bone and promotes osteoblast/odontoblast differentiation. ADHR is caused by increased FGF23 production or persistence. 6,114 All three of these hypophosphatemic disorders are associated with increased FGF23 activity, 46,112,118 impaired renal reabsorption of phosphate, and aberrant regulation of 1,25(OH)2D3 production. The hypophosphatemia is followed by defective calcification of cartilage and bone leading to rickets and osteomalacia. 15 Elevated FGF23 activity also occurs with paraneoplastic overproduction of the protein in tumor-induced osteomalacia. 83
Mice with inactivating mutations in the gene Phex show an XLH-like phenotype,8,96 while deletion of the Dmp1 gene results in ARHR-like disease, 27 and degradation-preventing mutations of Fgf23 produce changes compatible with ADHR. 6,15,114 HYP mice have an inactivating deletion in the Phex gene, 8,96 and the elevated circulating levels of FGF23 in these mice 95 lead to hypophosphatemia, impaired vitamin D metabolism, and hypomineralization of bone and dentin. 25 Similarly, Dmp1 deficiency in mice results in increased osteocyte production and elevated circulating levels of FGF23. 14,67 In addition, increased acidic serine aspartate-rich MEPE-associated motif (ASARM) peptides cause the mineralization defects in HYP mice. 20,74 Thus, inactivating mutations of either Phex or Dmp1 produce similar intrinsic bone mineralization defects as well as increased FGF23 expression by osteocytes. Comparative analysis of the Phex-mutant HYP mouse and Dmp1 null mouse showed that PHEX and DMP1 both regulate bone mineralization by inhibiting FGF23 production. 60 Consequently, the loss of function of either DMP1 or PHEX results in increased release of FGF23 by osteocytes. 52
Except for amelogenesis imperfecta, the phenotype of Fam20c –/– mice closely resembles those of HYP and Dmp1 null mice. All three of these lines show striking defects in dentin and rachitic changes in bone 27,119 accompanied by hypophosphatemia and elevated levels of FGF23. 26,54 Until now, mutations in only four genes (PHEX, FGF23, DMP1, and ENPP1) had been definitively linked to elevated levels of FGF23 and hypophosphatemic rickets. 55,79 Taken together, our findings strongly suggest that FAM20C should be considered an additional factor that influences biomineralization of dentin and bone by modulating levels of the phosphatonin FGF23.
Mouse models have been invaluable in advancing our understanding of biomineralization processes, and much has been learned by elucidating the molecular basis for changes in mineralized tissues using genetically engineered mice. A great advantage of mouse models is the availability of transgenic and other genetically engineered mouse lines to facilitate dissection of the precise molecular and pathogenetic mechanisms involved in mineralization. Our findings indicate that both FAM20A and FAM20C have critical (and most likely local) roles in forming the normal structure and mineralization of the extracellular enamel matrix. A variety of genes, including AMELX, ENAM, DLX3, FAM83H, MMP-20, KLK4, and WDR72, have been implicated in the development of AI in humans. 103 However, mutations identified within these genes account for less than half of all cases of amelogenesis imperfecta. 103 Our findings suggest that specific alleles of FAM20A and FAM20C may be responsible for some cases of AI in humans. In addition, we found that FAM20A is an important systemic inhibitor of subintimal mineralization of muscular arteries, possibly acting as a hormone in controlling the production and/or release of other mineralization inhibitors. The lethality observed in Fam20b –/– embryos supports a critical role for this gene in developmental processes. The changes observed in Fam20c –/– mice were typical of hypophosphatemic rickets and suggest a role for FAM20C in modulating FGF23 activity. Future additional studies on FAM20 family proteins should prove invaluable in obtaining a better understanding of the physiological and pathogenetic pathways involved in the mineralization of bones, teeth, and other tissues.
Footnotes
Acknowledgements
We thank June Wingert and Kathy Henze for their invaluable skills and efforts in providing necropsy and histology support. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported financially by Lexicon Pharmaceuticals, Inc., The Woodlands, TX 77381.