
Abstract
A diffuse, chronic, superficial neocortical degeneration that resulted in atrophy was detected in five 1 to 2-year-old-dogs. Presenting neurologic signs included ataxia, dysphagia, blindness, and mentation changes. Magnetic resonance imaging on brains from 2 dogs demonstrated severe bilateral cerebrocortical atrophy and enlarged lateral and third ventricles. Grossly, multifocal, bilaterally symmetrical, extensive areas of neocortical brownish discoloration associated with atrophy of gyri and sulcal widening were recorded in the dorsal and lateral cerebral hemispheres in 3 dogs. Microscopically, in all dogs there was subacute to chronic superficial neocortical degeneration affecting all cerebral lobes, ranging from loss of the molecular layer to less frequent larger and deeper cavitations of variable size. Clinical signs probably resulted from a combination of primary neocortical degeneration and secondary degeneration in the corticobulbar and corticospinal tracts. The distribution pattern of gross and histologic cerebrocortical lesions suggests that this is a novel degenerative canine cerebral disease.
Cerebrocortical necrosis and atrophy in dogs have been diagnosed as idiopathic 8,13 or associated with lead poisoning, 17 cyanide poisoning, 9 thiamine deficiency, 15 cardiac arrest, 14 hypoglycaemia, 10 cranial trauma, 12 following seizures, 2,11 or as a component of potentially hereditary degenerative central nervous system (CNS) disease. 1,3,4,6 Typically, neuronal degeneration and loss involve several laminae of the neocortex, sometimes resulting in full-thickness necrosis. In contrast, the idiopathic syndrome that we report here is characterized by very superficial (but occasionally deeper) neocortical degeneration and atrophy, encountered in 5 dogs in North America and the United Kingdom.
Materials and Methods
Four cases were in the personal collection of B.A.S., acquired from submissions to Cornell University College of Veterinary Medicine, Ithaca, New York (1982–2006). A fifth case was submitted to the necropsy service of the Royal Veterinary College, London, in 2012 (Table 1).
Signalment, Year Examined, Neurologic Abnormalities, and Organs Examined Postmortem of Affected Cases.a

aCase Nos. 4 and 5 had magnetic resonance imaging evaluation performed; case Nos. 1–3 did not.
Clinical Histories and Examination
Case no. 1, a 2-year-old intact female Scottish deerhound, was electively euthanized following a 3-month history of progressive neurologic abnormalities, including staggering gait, hyperextension of the thoracic limbs when walking, reduced proprioception especially on the right side, knuckling of left thoracic limb, and often behaving as if blind. The dog developed difficulty eating and drinking 2 weeks prior to elective euthanasia
Case no. 2, a 1-year-old male neutered Great Dane, presented with a 4-week history of gradual onset of neurologic abnormalities. He was first noted to have problems getting up and down stairs; over the following 3 weeks, he became progressively more confused and ataxic. On clinical examination, he was found to be hyperesthetic with loss of proprioception, mild ataxia, difficulty in prehending food, and intermittent partial focal seizures involving the jaw and neck. Elective euthanasia was performed.
No clinical history was available for case no. 3.
Case no. 4, a 2-year-old female neutered Toy Poodle, presented with a history of progressive neurologic signs of approximately 1-year duration. Signs ranged from loss of vision and hearing, self-mutilation, severe obtundation, and severe aggression. Episodes lasted approximately 5 minutes and had increased to 1 or 2 episodes per day. On clinical examination, she was depressed and hyperesthetic, reacting violently to being touched. No menace response, pupillary light response, or reaction to noise was detected. No hopping or patellar reflexes were elicited. Magnetic resonance imaging (MRI) of the brain was performed, but details of the sequences are not available. Elective euthanasia was performed.
Case no. 5, a 1.5-year-old female Irish Wolfhound, was electively euthanized following a 2-month history of progressive ataxia, hypermetria, blindness, and behavioral changes.
Magnetic resonance images were obtained with a 1.5-Tesla magnet (Philips Acheiva). The following sequences were acquired: T2-weighted, sagittal, transverse; T1 inversion recovery, transverse; T1 weighted, transverse, dorsal (pre- and postcontrast); and fluid-attenuated inversion recovery (FLAIR), transverse. For summaries of cases, see Table 1.
Necropsy and Histopathology
Case Nos. 2 and 4 had a complete necropsy; brain and cervical spinal cord were submitted in case Nos. 1 and 5; only the brain was submitted for case No. 3. Brains and other tissue samples were immersion fixed in 10% neutral buffered formalin, and selected samples were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin for initial microscopic evaluation. In case Nos. 3 and 5, additional stains used were reticulin, Masson’s trichrome, and Perls Prussian blue. Formalin-fixed, paraffin-embedded sections from case Nos. 3 and 5 were mounted on positively charged slides (Superfrost Plus, Menzl-Glaser) and immunolabeled for reactivity to glial fibrillary acidic protein (GFAP; monoclonal antibody, Leica), neuron-specific enolase (monoclonal antibody, Leica), and vimentin (monoclonal mouse antibody, Dako) through a peroxidase-based method for indirect immunohistochemistry on a Leica Bondmax autostainer. Diaminobenzidine chromogen was used for visualization of bound antibodies. GFAP was used at 1:100 with epitope retrieval solution 1 (ER-1; low pH) pretreatment. Neuron-specific enolase was used at 1:200 with ER-1 pretreatment. Vimentin was used at 1:500 with ER-1 pretreatment. Appropriate positive and negative controls were included. Cerebral tissue from case No. 5 was formalin fixed and subsequently refixed in glutaraldehyde, postfixed in osmium tetraoxide, and processed for transmission electron microscopy.
Results
Hematology and Biochemistry
Biochemistry and hematology results were available for dogs Nos. 2, 4 and 5, including serum resting and postprandial bile acids, and were within normal limits. Case No. 2 tested negative for serum lead. Thiamine levels of 72.0 ng/ml (reference range, 46–112 ng/ml) were detected in case No. 5.
Serology and Polymerase Chain Reaction
No antibodies to Ehrlichia canis, Rickettsia rickettsii, Neospora caninum, and Toxoplasma gondii agents were detected in serum from case No. 2.
Real-time polymerase chain reaction testing on cerebrospinal fluid (CSF) from case No. 5 for canine distemper virus, N. caninum, and T. gondii was negative.
CSF from case No. 2 had abnormalities, described as slightly elevated leukocytes consisting mainly of macrophages and a few lymphocytes and normal protein content. Case No. 5 had increased proteins and a mild eosinophilic pleocytosis.
Magnetic Resonance Imaging
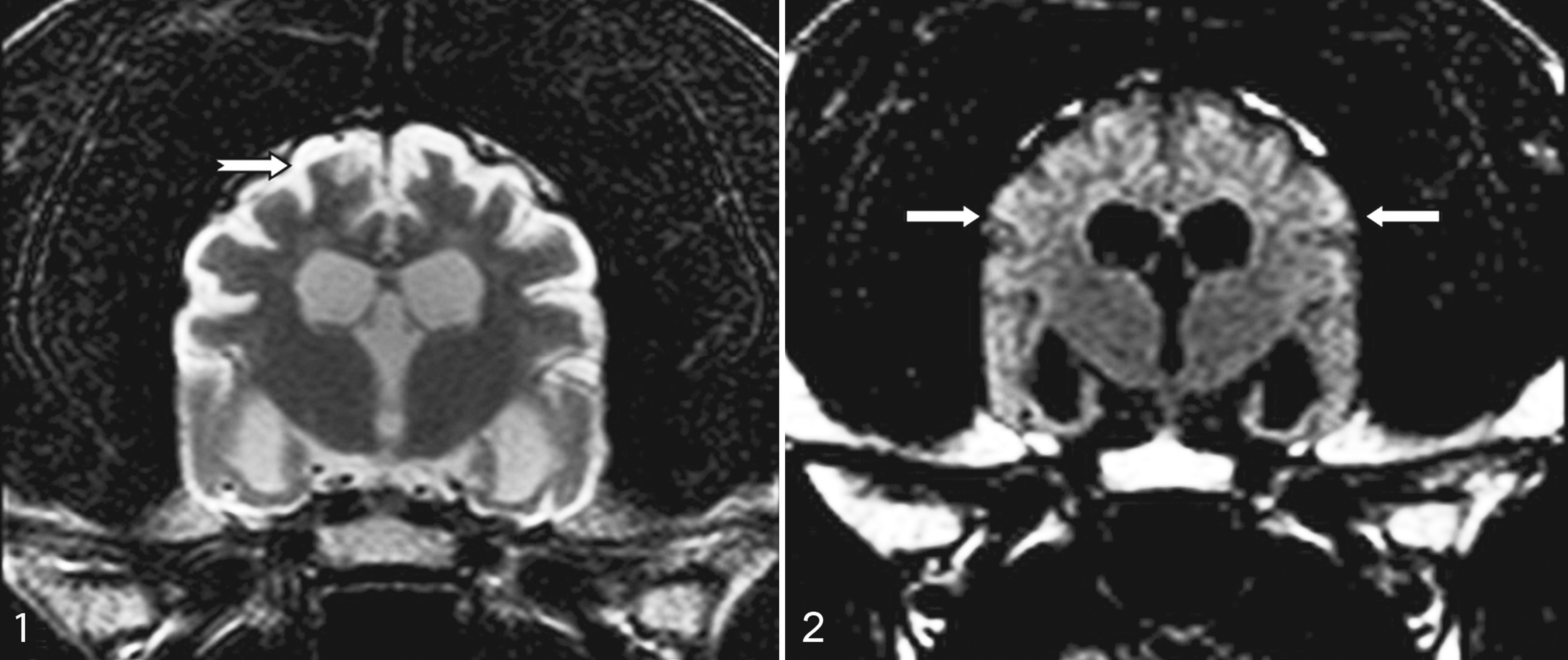
MRI examination was performed on case Nos. 4 and 5. Details are available only for case No. 5 (Figs. 1, 2), in which the T2-weighted images revealed hyperintense signal and widening of the subarachnoid space overlying the gyri and sulci of the frontal, temporal, and occipital lobes. In a similar distribution, the T1-weighted and FLAIR images revealed a hypointense (isointense to the CSF) signal at this level. A thin hyperintense signal rim in the cortical gray matter was present in the FLAIR sequences (laminar necrosis?). The thalamus (and interthalamic adhesion) was severely reduced in size, and enlargement of the lateral and third ventricles was present. The cerebellum did not show structural abnormalities. Postcontrast images revealed patchy irregular uptake of the neocortical meninges. The images were suggestive of cortical atrophy/necrosis and atrophy of the corona radiata, thalamus, and brainstem. In summary, MRI in dog Nos. 4 and 5 revealed severe bilateral cerebrocortical atrophy.
Pathologic Findings
Significant gross and histologic lesions were limited to the CNS in each case.
Macroscopic Lesions
In case No. 1, diffuse, markedly reduced neocortical gray matter was noted bilaterally, affecting the frontal and parietal lobes. A diffusely reddish appearance to cortical gray matter was noted in cross sections of cerebrum.
In case No. 2, on transverse sections of brain, marked bilaterally symmetrical atrophy affected the neocortex within the frontal, parietal, and temporal lobes of the rostral two-thirds of the cerebral hemispheres. This lesion was most extensive in the dorsal and dorsolateral gyri. The piriform lobes were atrophied and flattened with yellow discoloration. Dense white discoloration was noted of the tracts comprising the crus cerebri, longitudinal fibers of the pons, pyramids, and the pyramidal decussation. A round white spot was present bilaterally in the lateral funiculi in the position of the lateral corticospinal tracts of the cervical spinal cord.
No gross report was available for case No. 3. In case No. 4, no gross abnormalities were observed.
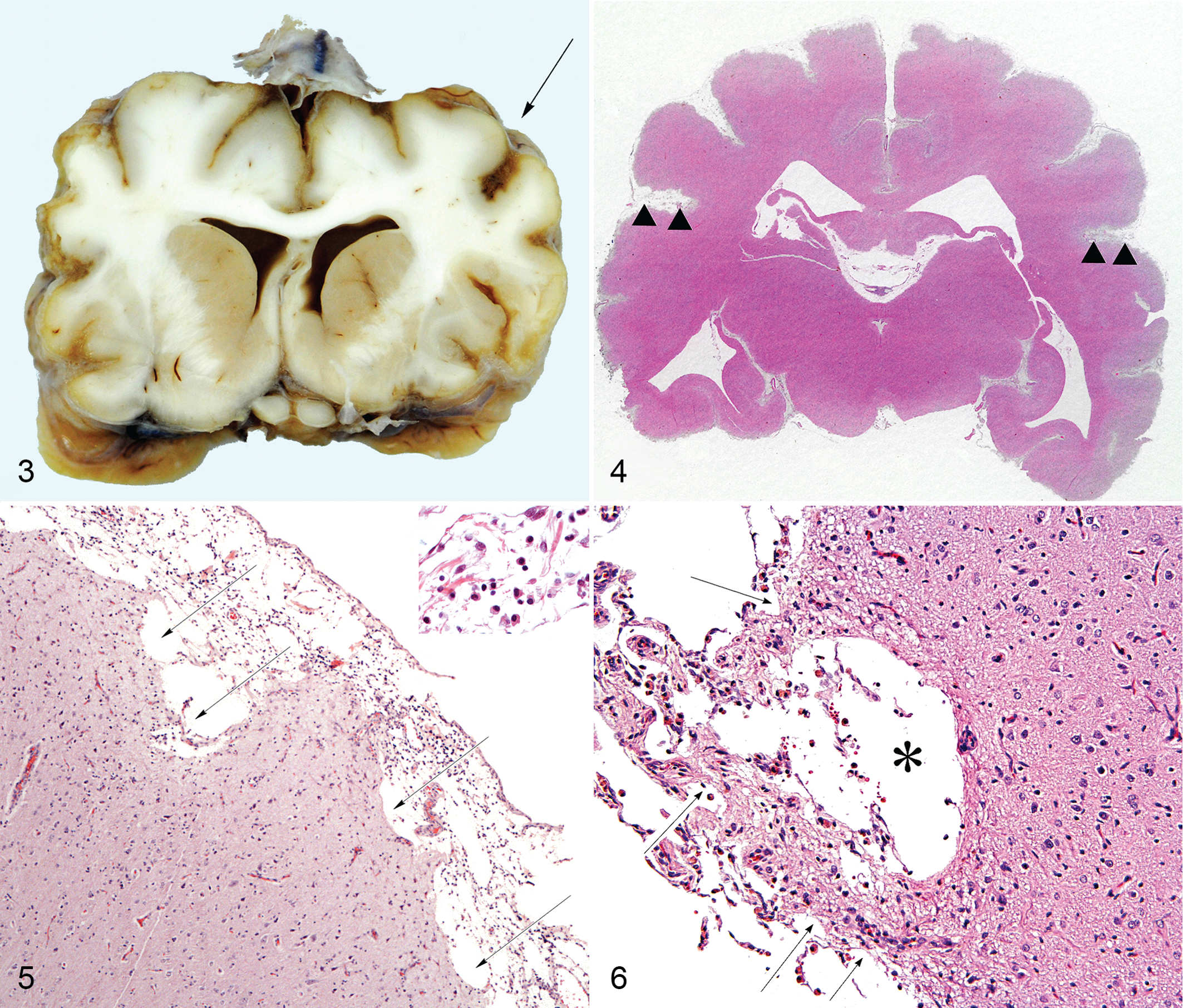
In case No. 5, multiple firm adhesions were noted between the cerebral hemispheres and the overlying dura mater throughout the cerebrum. A subtle patchy surface irregularity to the rostral two-thirds of the neocortex was noted, most extensively in the dorsal and dorsolateral gyri. On transverse sections of the brain, bilaterally symmetrical atrophy and patchy yellow discoloration of the neocortical gray matter of the parietal and temporal lobes were observed (Fig. 3). The lesions were most extensive in the dorsal and dorsolateral gyri.
Microscopic Lesions
Limited numbers of histology slides were available for examination in each case. Brain (and, when available, spinal cord) lesions in all cases were of a similar nature and are presented as a summary.
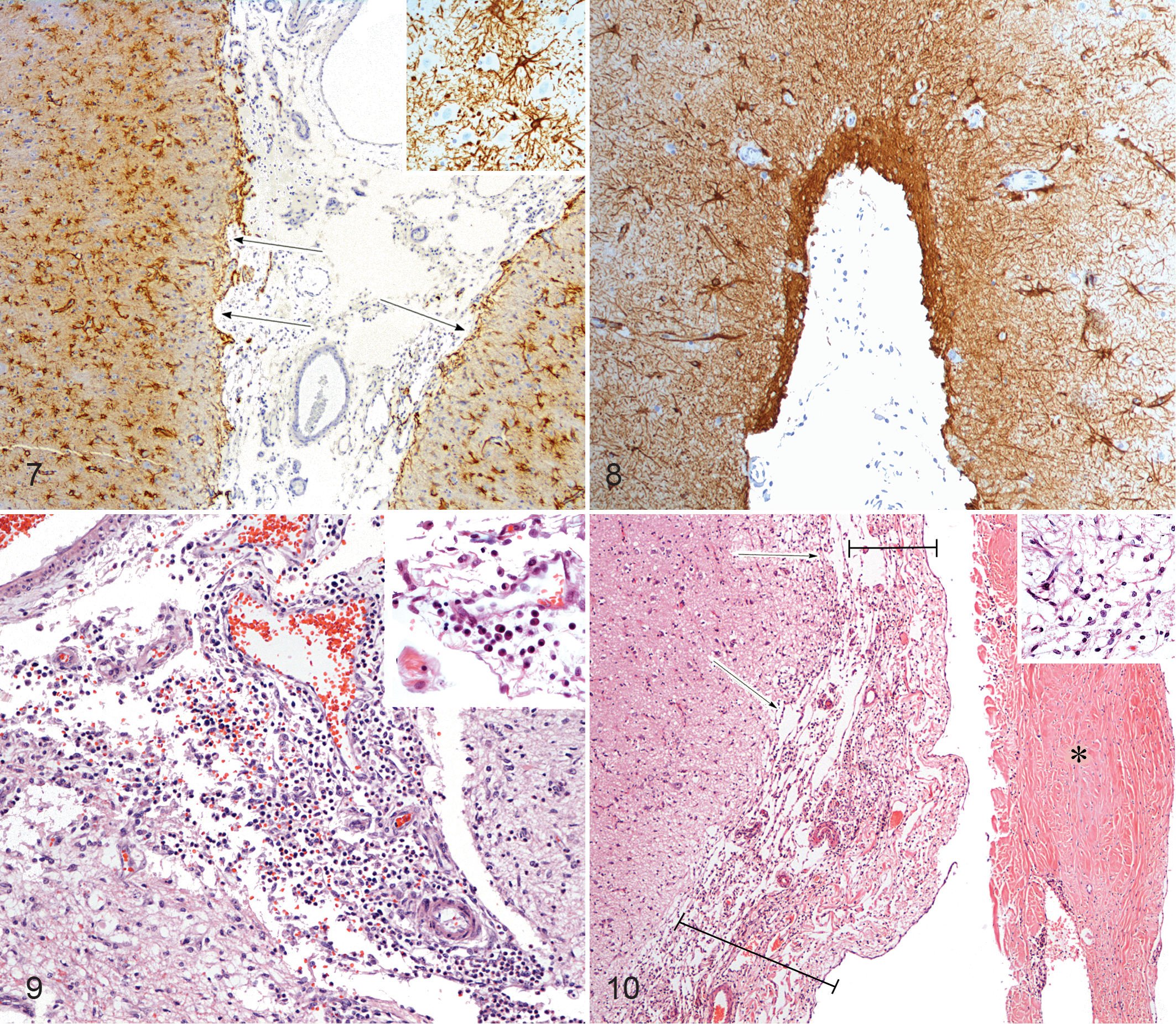
Approximately bilaterally symmetrical degenerative lesions diffusely affected the cerebral neocortex from the frontal to occipital lobes. These changes affected all surfaces of the cerebrum but most consistently the dorsal and lateral surfaces. Cerebrocortical degeneration and atrophy were sufficient to produce widespread marked widening of the sulci (Fig. 4). The normally smooth, convex cerebrocortical surface was undulating due to superficial tissue loss, producing scalloping or deeper cavitations, which extended into the molecular layer and sometimes further (Figs. 5, 6). Areas of neuropil degeneration were pallid and multifocally showed marked postnecrotic thinning or loss of the molecular layer (lamina 1) and occasional necrosis and loss of the most superficial neurones. Affected neocortex showed marked superficial astrogliosis—astrocytes appeared activated with enlarged nuclei and expanded cytoplasm. Such reactive astrocytes were found at all levels of the superficial to middle laminae and in denser aggregates involving the junction of the white matter. Astrogliosis was evident in GFAP (Fig. 7) and vimentin stains, mainly in superficial layers with middle to deeper layers largely unaffected, although deeper layers were notably affected in case No. 5. Mild neovascularization (capillaries and small arterioles) was frequently observed in areas of cavitation. A minimal to mild diffuse leptomeningeal and subarachnoid infiltrate of lymphocytes, plasma cells, and occasional neutrophils was often observed (Fig. 9), and perivascular cuffing was observed within the neocortex in case No. 5. Hemosiderin-laden macrophages were observed throughout the meninges in areas of neocortical necrosis in all cases in low to high numbers. Mature, debris-laden gitter cells within the areas of neocortical loss were only occasional, attesting to the chronicity of the cerebral injury. Leptomeningeal fibroplasia with moderate collagen deposition and increased vasculature was seen in case Nos. 3, 4, and 5 (Fig. 10).
Involvement of areas of the brain beyond the neocortex was seen occasionally. More acute lesions were observed in the hippocampal gray matter of case No. 3 and in the region of the occipital cortex of case No. 5 where prominent neutrophilic aggregates were noted in the leptomeninges, surrounding small-caliber leptomeningeal vessels lined by prominent endothelial cells and scattered in areas of necrosis.
In case No. 5, focally extensive necrosis of gray matter of the lateral aspect of the pons was characterized by loosening and rarefaction. Within the mesencephalon, diffuse subtle mild rarefaction of the superficial parenchyma with mild gliosis was noted. Similar lesions were observed affecting the gray matter on the surface of the hypothalamus.
Gray matter changes were not observed within sections of medulla oblongata and cerebellum, which were available for case Nos. 2, 3, and 5.
White matter degeneration characterized by myelin sheath ballooning, some loss of axons, rare intramyelinic macrophages, and astrogliosis was present within the corpus callosum, internal capsule, periventricular white matter, and bilaterally in the crus cerebri, longitudinal fibers of the pons, and the pyramids. This was present also in the lateral corticospinal tract in the cervical spinal cord of case Nos. 1, 2, and 5.
Discussion
We describe a novel but rare incapacitating and progressively worsening degenerative disorder of the CNS affecting young adult female and male dogs (3 were female [1 unknown sex]) presenting between 1 and 2 years of age that we encountered in the United States and the United Kingdom. Two were Irish Wolfhounds and one was a Scottish Wolfhound, raising the possibility of an hereditary basis. Onset of neurologic deficits was insidious and progressive, ranging from 1 month to 1 year. Neurologic signs included general proprioceptive and cerebellar ataxia, upper motor neuron paresis, blindness, and behavioral abnormalities. In addition to the 5 cases described, a further US case with strikingly similar lesions was assessed; however, signalment and full case history retrieval was not available, and so it was not included in this report.
Clinical neurologic signs in these dogs indicated multifocal to diffuse CNS disease with likely involvement of the prosencephalon (seizures, altered sensorium, central visual pathway) and caudal fossa/spinal cord (difficulty in prehension, general proprioceptive ataxia, and upper motor neuron paresis). Potential multifocal to widespread CNS disorders that were variously explored included evaluations for infectious causes of meningoencephalomyelitis, such as canine distemper, N. caninum, Neorickettsia, and encephalopathies, including lead poisoning, thiamine deficiency, and portosystemic shunting.
Two dogs received MRI evaluations, which identified the resulting neocortical loss and subarachnoid expansion and will play an important role in the diagnosis of any further cases.
The pattern of superficial cortical degeneration that we report is very unusual and differs from that commonly observed in cerebrocortical necrosis in dogs (or other species). Canine cerebrocortical necrosis is typically a sporadic condition occurring either alone or combined with gray matter lesions at other sites within the brain. 4 The cause is sometimes unknown 2,8 or follows cyanide poisoning, 9 hypoglycaemia, 10 trauma, 12 prolonged hypoxia following cardiac arrest, 14 lead poisoning, 17 and thiamine deficiency. 15 Cerebrocortical necrosis is also reported in disease processes, such as atherosclerosis, thromboembolism, meningitis, 2 infectious canine hepatitis, canine distemper encephalitis, 8 and gastroenteritis 7 and in congenital cellular metabolic disorders, as reported in the encephalopathy of Alaskan Huskies and Yorkshire Terriers. 1,4
In contrast to the lesions reported here, in which necrosis affected mainly superficial cerebrocortical laminae, canine cerebrocortical necrosis in previously described instances affected deeper laminae. 8 –10,12,17 In 1 case of prolonged hypoxia, all layers of the cerebral cortex were affected. 14 In Alaskan Husky encephalopathy, necrosis variably affected superficial to deep layers of the cerebral cortical laminae with lesions occurring mainly at the base of the sulci. 4 In the cases reported here, gray matter necrosis and loss were specifically limited to the superficial neocortex, mainly the molecular layer (lamina 1) affecting the neuropil with occasional neuronal necrosis. Lesions were present in the sulci and gyri.
Subtle gray matter rarefaction, necrosis, and gliosis were observed in areas of the hypothalamus, mesencephalon, and pons from case Nos. 3 and 5, suggesting that lesions are not limited to neocortical gray matter in these cases. This raises the possibility of the presence of a CSF toxin; however, a toxin within the CSF would have access to the ventricles, and no evidence of ependymal necrosis was noted.
We interpret the widespread superficial neocortical degeneration and loss as the primary lesions with bilateral white matter degeneration a consequence of diffuse neocortical loss. This degeneration observed grossly in case No. 2 and otherwise microscopically in the midbrain, pons, medulla, and spinal cord is bilateral and symmetrical and anatomically consistent with the descending corticobulbar and corticospinal tracts. These changes reflect degeneration of upper motor neuron axons and concurrently their myelin sheaths or transsynaptic interneuron loss. The severity of the white matter degeneration reflects the widespread bilateral neocortical lesion and the chronicity of these changes but is surprising given their superficial nature.
Bilateral and symmetrical CNS lesions in either gray or white matter or affecting both simultaneously are generally caused by aberrations in global vascular perfusion (hypoxia-ischemia) preferentially affecting areas of highest sensitivity or associated with metabolic dysfunction due to neurodegenerative disorders or toxicoses.
Global brain ischemia as can occur in cardiac arrest 14 and anesthetic accidents targets areas of the neocortex, hippocampus, and cerebellum. Cerebral circulatory disturbances in humans more often occur in gray matter at the base of sulci rather than at the crest. 5 Our cases showed gray matter necrosis at the base and crest of sulci, affecting the outermost cortical laminae with relative sparing of most laminae of neurones. Neocortical necrosis in human cases of cardiac arrest is most evident in the occipital and parietal lobes where the first and second laminae are usually preserved. In canine cases of cardiac arrest, all layers of the cerebral cortex are involved. 14 Furthermore, we are unaware of any antecedent events, such as cardiac arrest or general anesthesia, in these 5 relatively young dogs. In addition, the clinical signs in these dogs were progressive which would not be expected in most vascular disorders. The possibility of a hypoxic, late gestation or periparturient injury was entertained, however given the lack of neurological signs observed in these dogs as juveniles this was considered an unlikely etiology. Superficial neurodegeneration (in addition to gross infarction) is seen in some cases of feline ischemic encephalopathy caused by intracranial Cuterebra migration. 16 It is presumed that an excreted parasite product is neurotoxic. While this causes superficial erosive neocortical degeneration and associated gliosis, lesions are asymmetric and involve the ventricular system with ependymal loss (presumably, the putative toxin is spread in CSF); these changes are lacking in these dogs.
Superficial astrogliosis accompanied the disruption of the neocortex, well demonstrated with GFAP and to a lesser degree with vimentin stains, and was evident on ultrastructural examination. Sometimes astrocytic changes extended more deeply, most notably in case No. 5, involving the gray-white matter junction, suggesting deeper injury in these instances. Gitter cells were absent from the parenchyma, but some mononuclear macrophages contained hemosiderin, attesting to prior hemorrhage. High numbers of neutrophils and lesser numbers of histiocytes and occasional lymphocytes were observed in acutely affected regions in case Nos. 3 and 5 within the leptomeninges and subarachnoid space. This is interpreted as secondary to tissue necrosis. Leptomeningeal fibrosis was sometimes noted also.
Intoxication by an exogenous agent was considered unlikely, given the novel lesions, narrow age range of the affected dogs, and sporadic occurrence of cases (5 in approximately 30 years). Cerebrocortical lesions secondary to seizures have been described, 11 but seizure activity was reported in only 1 of the 5 dogs in this report. Cortical necrosis, likely seizure induced, is seen in some cases of canine distemper encephalitis selectively affecting the pyriform lobe and the hippocampus, 2 and it affects middle to deeper laminae of neurons.
As 2 dogs were of Irish Wolfhound origin and one of Scottish Wolfhound origin (related and not exceptionally common breeds), a hereditary basis for the cases was entertained. Canine CNS disorders of unknown causes that are potentially inherited and characterized by symmetrical gray matter degeneration and necrosis with neuronal preservation include a neurodegenerative condition in Australian Cattle dogs, 3 familial cerebellar ataxia with hydrocephalus in Bull Mastiffs, 6 and encephalopathy of Alaskan Huskies and Yorkshire Terriers. 1,4 While all these disorders present in early life, they are quite distinct neuropathologically from the syndrome that we describe herein for which the cause and pathogenesis remain to be elucidated.
Footnotes
Acknowledgements
We acknowledge the assistance of Professor Ken Smith, Department of Pathology and Pathogen Biology, Royal Veterinary College, London, in providing support for this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.