
Abstract
A novel leukoencephalomyelopathy was identified in 73 mature male and female large captive felids between 1994 and 2005. While the majority of identified cases occurred in cheetahs (Acinonyx jubatus), the disease was also found in members of 2 other subfamilies of Felidae: 1 generic tiger (Panthera tigris) and 2 Florida panthers (Puma concolor coryi). The median age at time of death was 12 years, and all but 1 cheetah were housed in the United States. Characteristic clinical history included progressive loss of vision leading to blindness, disorientation, and/or difficulty eating. Neurologic deficits progressed at a variable rate over days to years. Mild to severe bilateral degenerative lesions were present in the cerebral white matter and variably and to a lesser degree in the white matter of the brain stem and spinal cord. Astrocytosis and swelling of myelin sheaths progressed to total white matter degeneration and cavitation. Large, bizarre reactive astrocytes are a consistent histopathologic feature of this condition. The cause of the severe white matter degeneration in these captive felids remains unknown; the lesions were not typical of any known neurotoxicoses, direct effects of or reactions to infectious diseases, or nutritional deficiencies. Leukoencephalomyelopathy was identified in 70 cheetahs, 1 tiger, and 2 panthers over an 11-year period, and to our knowledge, cases have ceased without planned intervention. Given what is known about the epidemiology of the disease and morphology of the lesions, an environmental or husbandry-associated source of neurotoxicity is suspected.
Keywords
In large nondomestic felids, primary neurodegenerative disease confined to central nervous system (CNS) white matter is rare. Examples of reported conditions include a clinical ataxia syndrome of unknown origin described in juvenile and adult European cheetahs and characterized by spinal cord white matter degeneration 33 and degenerative myelopathies secondary to deficiencies in either copper 36 or vitamin A. 19,26
In 1994, a 13-year-old captive Florida panther (Puma concolor coryi) was euthanized after developing severe neurologic dysfunction. Postmortem examination of the brain revealed a novel bilateral degenerative lesion located primarily in the cerebral subcortical white matter; the first recognized case of large felid leukoencephalomyelopathy (LFL). This case was followed in 1996 by diagnosis of the same lesion in a captive cheetah (Acinonyx jubatus) through its inclusion in a cooperative program initiated in 1988 by the Association of Zoos and Aquariums (AZA). To ensure careful genetic management and because diseases such as hepatic veno-occlusive disease, glomerulosclerosis, and gastritis were a significant impediment to maintaining self-sustaining captive cheetah populations, a health surveillance program with a pathology survey was developed for captive cheetahs by the AZA’s Cheetah Species Survival Plan (SSP). 21 Through this survey, 36% of submissions between 1996 and 2005 were diagnosed with LFL, as were an additional captive Florida panther and a captive tiger. To our knowledge, the last confirmed case of LFL was diagnosed in 2005. The purpose of this report is to present the clinical and pathologic features of this novel felid leukoencephalomyelopathy and discuss aspects of its epidemiology as investigated before its apparent disappearance.
Materials and Methods
Formalin-fixed brains or histologic brain sections as well as samples of all major organs from 198 captive cheetahs that died between January 1996 and December 2005 were received from zoos within the United States as part of the Cheetah SSP pathology survey. Spinal cords submitted in a subset (n = 10) of the cheetah submissions were also examined. For animals that died during this time frame, clinical information, including the nature and duration of neurologic signs, was specifically requested. Over the same period, brains from a small number of other felid species (Florida panther, Puma concolor coryi, n = 2; tiger, Panthera tigris, n = 1) with clinical presentations suggestive of LFL were also submitted as candidate cases with complete medical records. Archived brain sections from the Cheetah SSP pathology survey from animals that died before 1996 (n = 77), as well as samples obtained from 2006–2012 (n = 91), were reexamined for LFL lesions. CNS tissues were sectioned transversely every 5 mm, examined grossly, and all visible lesions trimmed for histopathology. Representative sections of the cerebrum (frontal, temporal, parietal, and occipital lobes, including hippocampus), brainstem, cerebellum, and spinal cord (as available) were also sampled. Tissues were processed routinely, embedded in paraffin, sectioned at 4 to 6 μm and stained with hematoxylin and eosin. All microscopic lesions were anatomically and morphologically characterized, and complete maps relating anatomic distribution to severity of lesions were produced for 32 randomly selected cases. Lesions were considered mild if they had prominent gemistocytic astrocytes but minimal to no myelin or axonal loss; moderate lesions had more extensive astrocytosis with white matter degeneration; severe lesions had marked astrocytosis and extensive white matter degeneration with rarefaction and cavitation.
Cerebral samples from 20 cheetahs with mild, moderate, and severe histopathologic lesions were selected for special staining procedures, including Luxol fast blue (LFB)–periodic acid–Schiff (PAS) and Sevier-Munger or Bielschowsky stain, to assess myelin and axon loss, respectively. Astrocytosis was assessed in the selected cases by glial fibrillary acidic protein (GFAP) with streptavidin-biotin immunohistochemistry employing a rabbit polyclonal antibody at 1:3000 (DAKO, Carpinteria, CA) without pretreatment of slides. Glutaraldehyde or formalin-fixed samples of corona radiata from 16 affected cheetahs representing the spectrum of mild to severe lesions and 1 unaffected age-matched (11-year-old) cheetah were selected for electron microscopy. The tissues were postfixed in 2% OsO4, embedded in Epon, and evaluated as 1-μm-thick sections stained with toluidine blue. From selected areas, ultrathin sections stained with lead citrate and uranyl acetate were examined on a Phillips 301 electron microscope (Philips Electronics North America Corporation, Andover, MA).
Results
Animal Population
In sum, 279 cheetahs died in AZA member institutions between January 1996 and December 2005. Tissues from 198 of these 279 cheetah were submitted to the Cheetah SSP pathology survey. Of the 198 examined, 36% (n = 70) had some degree of LFL. No additional cases of LFL were identified in tissues submitted from 1988 to 1995 (n = 77) or since December 2005 (n = 91). There were, however, 11 cases since 2005 that, within white matter tracts, had slightly increased numbers of glial cells, edema, and gitter cells within a few dilated axon sheaths. These cases lacked prominent or atypical astrocytes, which were a hallmark of leukoencephalomyelopathy lesions and thus did not match the case definition. Additionally, no cavitation was noted in these cases. Complete pedigree analysis, conducted on all reported cases of LFL in cheetahs, indicated that the disease appeared in the cheetah lineage without following family lines and was most prevalent in 2 generations before an incremental decline beginning in 1998. The majority of animals were not diagnosed with LFL, and cases appear to have been random and ephemeral.
In addition to cheetahs, 2 Florida panthers—1 female aged 12 years and 1 male aged 13 years—were euthanized in 1994 and 1997, respectively, and diagnosed with LFL. Both panthers were housed at a facility that also had 9 affected captive cheetahs. Subsequent to these cases, in 2000, a single female tiger (Panthera tigris) aged 18 was euthanized at a facility with no other known cases of LFL and diagnosed with this disease.
Affected felids (70 cheetahs, 2 panthers, 1 tiger) ranged from 7 to 18 years old (median and mean, 12 years) with only 7 animals aged younger than 10 years at the time of postmortem examination. Males and females were equally affected (females, n = 38; males, n = 35). Animals were housed at 29 zoological facilities throughout all geographic regions of the United States and a single location in the United Kingdom. Three US facilities with large cheetah collections housed 33 of the 73 (45%) affected animals. The temporal occurrence of case diagnoses (based on postmortem neuropathologic examination) peaked in 1998, with 22 new cases diagnosed in that year, and tapered until the final case was diagnosed in 2005 (Fig. 1), with reported clinical onset mirroring these data.
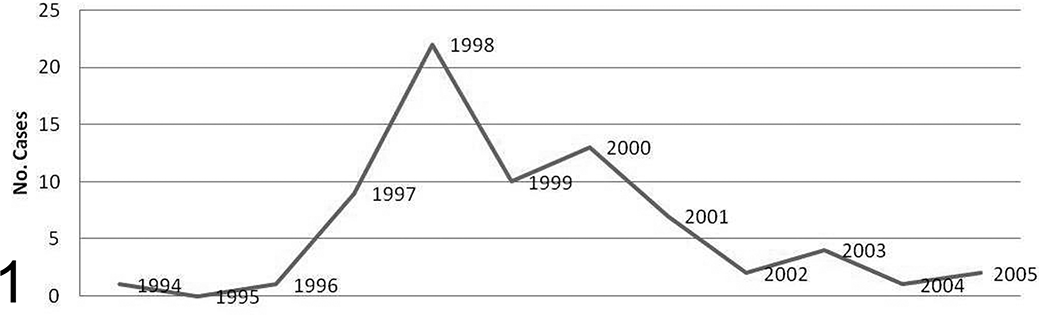
Temporal occurrence of postmortem diagnoses of large felid leukoencephalomyelopathy.
Clinical Summary
The index case was a 13-year-old male Florida panther that had been housed at the same location for a little over 8 years before the development of ataxia and suspected blindness. These clinical findings progressed for just over 1 month, with the addition of altered behavior, possible proprioceptive deficits, and, finally, seizures necessitating euthanasia in June 1994. Similar subtle signs of diminished vision began to appear in aged cheetahs at this and other facilities from late 1996. In general, affected large captive felids first lost the ability to visually follow keeper activities or locate food in enclosures. Affected animals had a blank stare, as if not fully cognizant of their surroundings, or appeared quieter than normal. Animals became progressively disoriented within familiar surroundings, walking into enclosure walls and misjudging the location of doorways. Affected cheetahs had difficulty eating, developed erratic head movements, and bit at food dishes and their own paws. Seizures occurred in some cases. The duration of clinical signs ranged from a few days to 2 years, typically ending with humane euthanasia. Clinical signs were exacerbated by any change in management, such as introduction of a new animal or moving the affected animal to a new enclosure. One of the first clinical observations made was an unusual response to anesthetic agents during induction and recovery. Inductions were typically prolonged and required higher doses of anesthetic. Recoveries were also prolonged and stormy, and neurologic clinical signs were more pronounced following recovery. Several animals became moribund during anesthesia, requiring euthanasia. Some affected cheetahs had no apparent clinical signs, including a few individuals with severe white matter loss found on postmortem examination after death due to other causes. Because cases were in large nondomestic felids, clinical neurologic examinations were limited and assessment largely observational. Degenerative changes occurred in white matter tracts associated with the central visual pathway, as well as the projection pathways for the upper motor neuron corticospinal pathway and conscious proprioception. Thus, the neurologic deficits may have resulted entirely from blindness or may have included some gait deficit from the involvement of the corticospinal and conscious proprioceptive systems. Once the clinical disorder began, in no cases did the neurologic signs resolve.
Imaging Findings
Both magnetic resonance (MR) and computed tomography imaging studies were used as diagnostic aids at some facilities, with T2-weighted MR images showing the greatest lesion detail and providing the most definitive antemortem diagnostic test. T2-weighted images were hyperintense in areas with white matter loss, highlighting the influx of fluid into these areas (Fig. 2). MR images clearly substantiated the bilateral symmetry of LFL and in a majority of cases showed dilation of the lateral ventricles. While interpreted as secondary hydrocephalus in many cases, there were some cases with enlarged ventricles that had relatively mild white matter lesions. One facility, having conducted MR imaging at multiple periods in the progression of some cases, reported that MR lesions could be subtle early in the disease process but would be very apparent later. T1-weighted MR images showed hypointense cerebral white matter (Fig. 3), and computed tomography studies identified hypodense cerebral white matter.
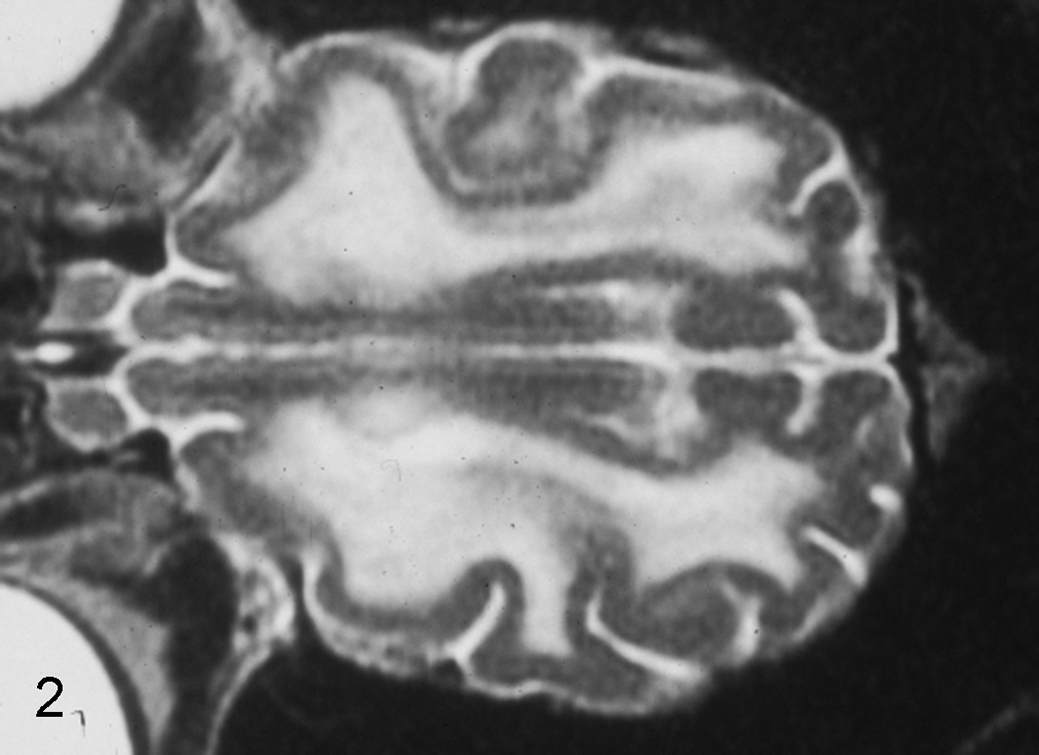
Brain, cheetah, magnetic resonance image. Lesions of large felid leukoencephalomyelopathy readily visible in a T2-weighted axial section as bilaterally symmetric hyperintensity in areas of severe myelin loss in the dorsal plane.
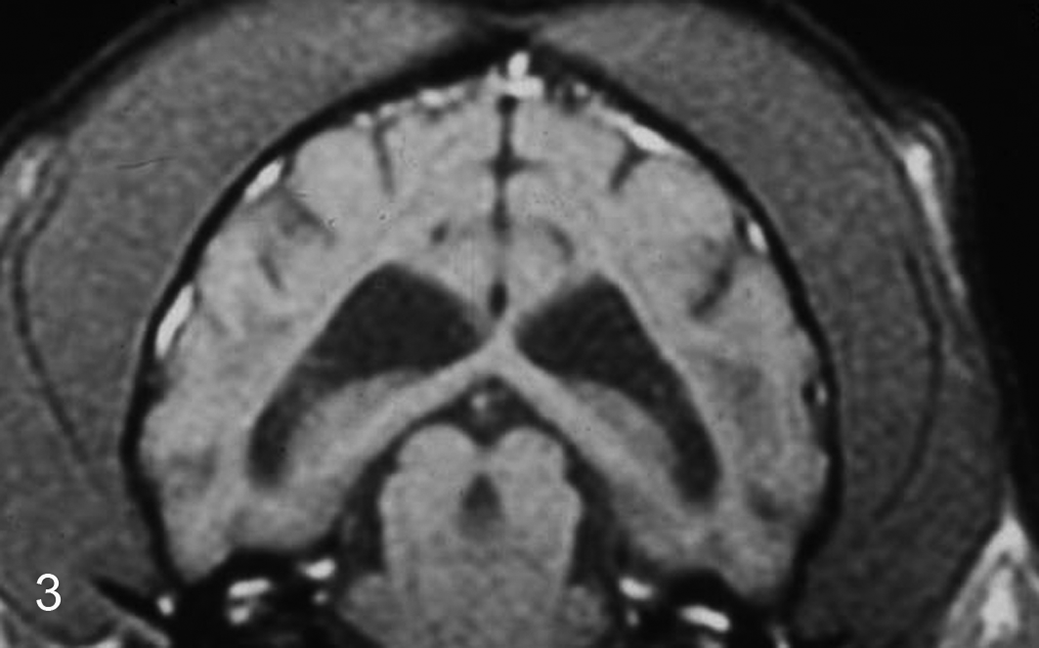
Brain, cheetah, magnetic resonance image. Lesions of large felid leukoencephalomyelopathy shown in a T1-weighted coronal section as bilaterally symmetric hypointensity in areas of myelin loss, most apparent in this image as gray areas located adjacent to the lateral ventricles. This image also shows dilation of lateral ventricles.
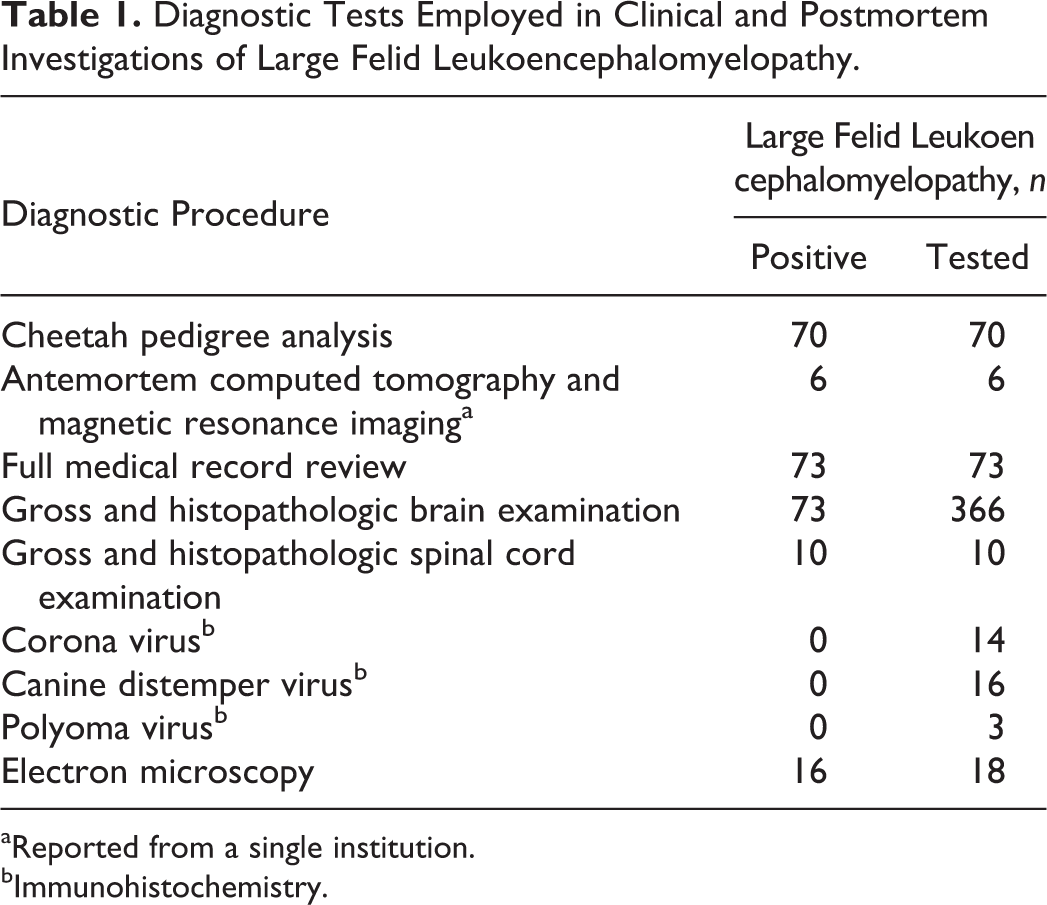
Diagnostic Tests Employed in Clinical and Postmortem Investigations of Large Felid Leukoencephalomyelopathy.
aReported from a single institution.
bImmunohistochemistry.
Gross and Microscopic CNS Pathology
In all cases, the principal lesions occurred in the cerebral white matter. Both in appearance and on palpation, the intact brain was normal, but on brain transverse sections, those cases with severe LFL had grossly evident bilateral areas of malacia, which appeared as graying and cavitation of white matter. To varying degrees the centrum semiovale and corona radiata in all cerebral lobes were affected. Marked enlargement of the lateral ventricles subsequent to the loss of periventricular white matter (secondary hydrocephalus) was also evident in severely affected animals.
Regardless of severity, white matter of the frontal lobe was consistently involved, and the centrum semiovale of this region typically showed the most severe changes (Figs. 4, 5). Lesions ranged from widespread involvement of cerebral white matter extending to the margins of the deep cortical gray matter to patchy areas of white matter degeneration and gliosis in the core of the corona radiations in gyri, in many cases sparing the adjacent centrum semiovale, though cavitation could be found in either of these white matter locations. Lesions tended to have a patchy distribution in the less severe cases but were bilateral and anatomically symmetrical. Severity tended to decrease in the internal capsule and in more caudal sites of white matter involvement, where gliosis could be found without overt white matter degeneration. Of the 32 fully mapped cases, 40% (n = 13) showed degenerative changes in white matter beyond the cerebral hemisphere in the following CNS regions: thalamus, crus cerebri, longitudinal fibers of the pons, cerebellum, pyramids, and the lateral corticospinal tracts in the lateral funiculi of the spinal cord. It is possible that with the exception of the cerebellum, all these lesions in the brainstem and spinal cord are secondary to the interruption of the projection pathways from the cerebral cortex within the white matter of the cerebral hemispheres. While myelin loss was variably noted in these regions, none had the severe changes and cavitation found in cortical white matter. Anatomic locations in which white matter degeneration and/or gliosis were never found include optic tracts, corpus callosum, fimbria of the hippocampus, and ventral funiculi of the spinal cord. Of the cases mapped, 25% (n = 8) were classified as having mild lesions, 34.4% (n = 11) as moderate, and 39.4% (n = 13) as severe. It should be emphasized that euthanasia and concurrent disease interrupted clinical progression of neurologic disease in most cases. One facility that housed a significant proportion of the affected cheetahs and both panthers reported that most cases could have lived with LFL for much longer than they did but that it became very difficult for animal caretakers to watch the animals deteriorate. In only a few cases where seizures were a prominent part of the clinical presentation was euthanasia absolutely necessary at this facility.
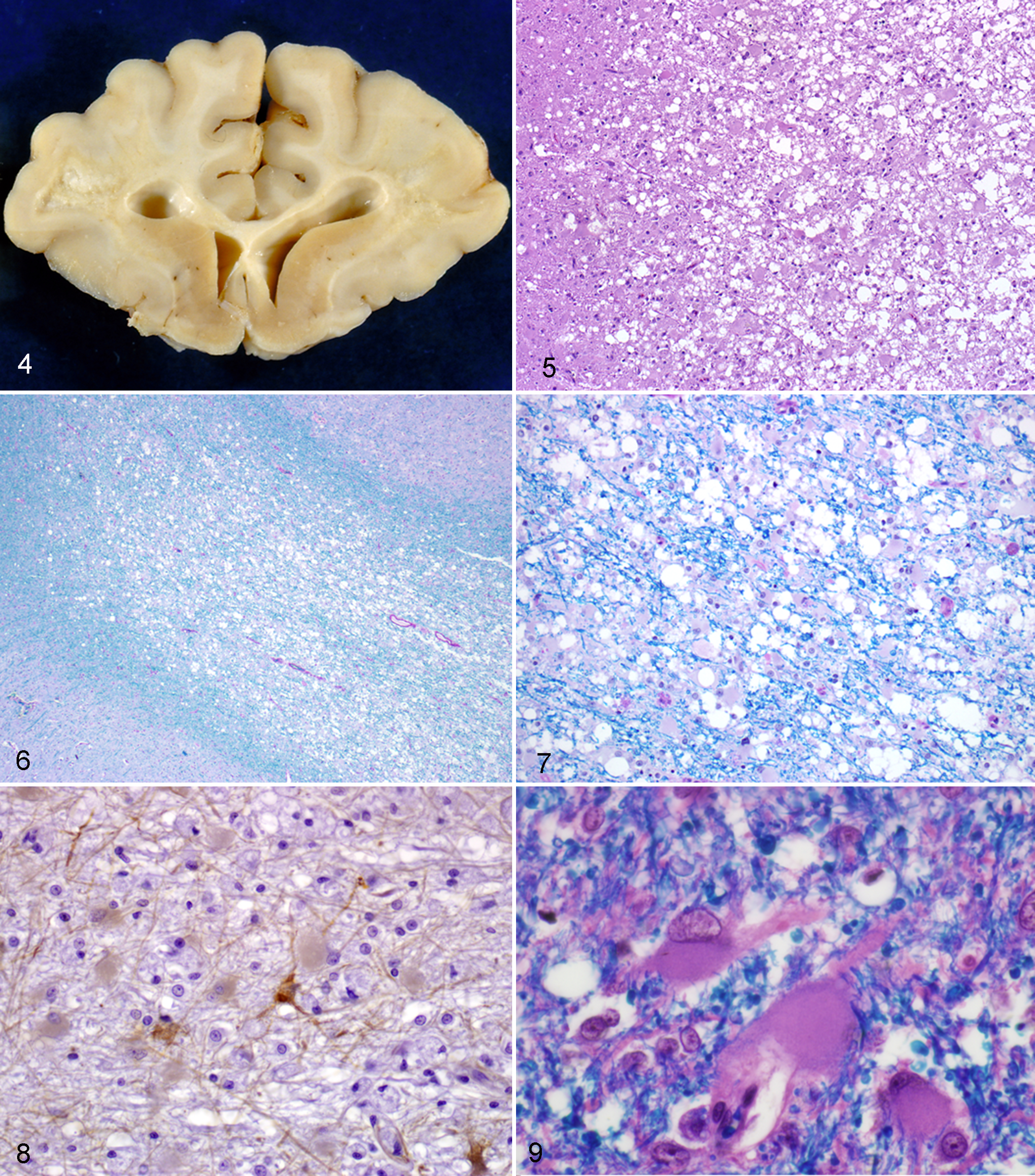
Microscopic features were most consistent with a chronic, ongoing degenerative process, and the extent of myelin and axon loss was clarified in LFB-PAS (Figs. 6, 7) and Bielschowsky stained sections. A striking feature of the lesions noted in affected brain and spinal cord white matter regardless of degree of white matter degeneration was the presence of an astrocytic population with exceptionally abundant, finely granular eosinophilic cytoplasm with vacuolation, reduced GFAP staining, karyomegaly, and multinucleation. In addition to this astrocytic appearance, lesions contained more typical-appearing gemistocytic astrocytes with abundant GFAP-positive cytoplasm and processes (Figs. 8, 9). In severe cases, most myelinated axons and interfascicular oligodendrocytes were replaced by loose networks and random plaques of astrocytes with both morphologies, capillary sized blood vessels, and interspersed gitter cells. In some severely affected animals, scattered foci of white matter and glial (astroglial and oligodendroglial) mineralization were noted. Gitter cells in white matter and surrounding blood vessels contained abundant myelin debris and other PAS-positive material. Approximately 50% of severely affected cases had perivascular lymphocyte cuffs near affected and within unaffected areas, and 2 cases had extension of lymphocytes into the degenerate white matter. Severely affected animals also had gemistocytic astrocytes in the deep laminae of the neocortex adjacent to corona radiata. Cerebral gray matter otherwise appeared normal.
Moderate lesions of LFL had abundant astrocytes as described and rarefaction of white matter due to loss or vacuolation of myelin sheaths. Most axons coursed through the lesion intact, suggesting that in less severe lesions, myelin loss preceded axonal destruction. In some moderate cases, large numbers of gitter cells were distributed within the parenchyma and in clusters around blood vessels, and focally swollen axons were present. Mild forms of LFL consisted principally of accumulations of conventional and atypical astrocytes, as described, in the white matter of the corona radiata with only minimal swelling or vacuolation of myelin sheaths and rare axon swelling.
Ultrastructural CNS Pathology
Ultrastructural lesions in the 16 cases examined consisted of degenerative changes in myelin sheaths and axons. These included unsheathed axons, empty myelin sheaths, and myelin sheaths in varying stages of disruption and collapse with astrocytic cytoplasmic processes extending between myelin lamina. The large bizarre astrocytes noted by light microscopy had deeply folded nuclei, finely granular cytoplasm, a paucity of cytoplasmic intermediate filaments, and no intermediate filament aggregates. Oligodendroglia appeared normal. Throughout all regions, mononuclear macrophages contained degraded myelin and amorphous debris in the cytoplasm. Blood vessels were encircled by pericytes or perivascular microglia containing abundant membrane-bound cytoplasmic debris. In areas of advanced white matter loss, numerous filament-rich astrocytic processes formed dense scars, within which occasional lipid droplets, macrophages, and rare remaining myelinated axons were present.
Concurrent Pathology
Most of the animals affected with LFL had coexisting conditions that are common in the captive cheetah population, including hepatic veno-occlusive disease, renal amyloidosis, glomerulosclerosis, Helicobacter-associated gastritis, and oxalate nephrosis. LFL was the principal reason for euthanasia in approximately 50% of the cases, while gastritis, hepatic, and renal disorders were the primary cause of clinical signs leading to death or euthanasia in the remainder.
Table 1 provides a summary of antemortem and postmortem diagnostics conducted through the period in which LFL was diagnosed.
Discussion
The clinical and pathologic features of LFL—a late-onset, progressive, often fatal leukoencephalomyelopathy of captive felids predominantly in the United States—are unlike those of any previously described disease in any species. This disease appeared in captive felids over an 11-year period and then disappeared from monitored populations. The salient clinical presentation in most cases was progressive loss of vision, advancing to blindness and seizures. MR imaging as well as gross, microscopic, and ultrastructural studies demonstrated progressive white matter degeneration, multifocal or diffuse, involving the CNS, bilaterally and extending from the frontal lobe, primarily along projection pathways, caudally to as far as the spinal cord in some cases. Lesions ranged from mild to severe, a spectrum of changes that may reflect individual animal differences and the length of the clinical course, as some cheetahs were euthanized after a few months, while others lived up to 2 years after onset of clinical signs. A prominent and unusual astrocytosis across cases was observed irrespective of severity and may suggest that damage to white matter astrocytes is a step in the pathogenesis of the lesion. 18,20
However, atypical astrocytes are sometimes observed in chronic gliosis in humans, and such changes (eg, degenerative nuclear atypia) must be distinguished from primary gliodegeneration. 6 While the low level of GFAP staining in the larger astrocytes may be due to the transience of GFAP upregulation, 20,25 consistent with the chronic, ongoing nature of LFL, the distinctive nuclear and cytoplasmic features of this morphologic subset of astrocytes was not characteristic of typical reactive astrocytosis. 20,24 In some cases, significant myelin degeneration, or overall myelin loss as noted on LFB stains, did not appear to be present in the face of florid astrogliosis. Thus, a proposed sequence of events may be astrocytic damage or derangement, followed by myelin degeneration and typical microglial responses and, finally, axonal loss and cavitation. Lesions lacked a consistent inflammatory component and showed bilateral symmetry, suggesting direct damage (ie, toxin) or a neurometabolic derangement as the initiating event. Selective vulnerability of different regions of the CNS is typical of neurotoxicity. 24 In these felids, the cerebral white matter appears to be the most vulnerable area for the presumptive neurotoxin. While LFL is clearly a distinct and newly identified neurologic disease, it does have features in common with other leukoencephalopathies. Fibrinoid leukodystrophy (Alexander disease) of humans and some animals is now known to be a primary astrocytic disease with mutations in the GFAP gene that lead to diffuse or focal demyelination. 15,32 While the distribution and appearance of Alexander disease bears some resemblance to LFL, Rosenthal fibers (a key histologic feature of Alexander disease) were not found in LFL, and the epidemiology and signalment of LFL set these diseases apart. Multiple sclerosis (MS) is the most common demyelinating condition of humans, and, like LFL, the cause is not known. MS is distinct from LFL in clinical onset (MS typically presents in early adult life) and in lesion distribution and appearance. Lesions of MS can appear throughout the CNS, are asymmetrical, and typically wax and wane over time. 18 In contrast, most lesions in individual felids with LFL appeared of similar duration throughout the brain and were largely symmetrical within the CNS white matter, suggesting a course of progressive degeneration from onset in LFL.
In Europe, degenerative lesions of the spinal cord white matter have been described in cheetah cubs and in 7- to 10-year-old adult cheetahs. 33,34 Clinical signs were referable to progressive spinal cord deficits and revealed degenerative changes in select white matter tracts. However, evidence of blindness was not reported, and cerebral white matter degeneration, the most characteristic lesion of LFL, was lacking. As with LFL, the cause of this neurodegenerative disorder in European cheetahs remains unknown. Similar to the adult cheetahs, 33 leukomyelopathies also occur in mature dogs, cats, and rats. Variably distributed perivascular lymphocytic cuffs were found in the CNS of some LFL cases. While this finding, in the context of the overall lesion, appears more likely incidental or indicative of an immune response to altered myelin antigens, infectious causes were considered in initial investigations, and evidence of viral infection was sought for several reasons. Chronic canine distemper virus causes demyelination and is known to affect large cats, 29 and corona viruses have been associated with demyelination in other species. Examples of the latter include presence of corona viruses in some MS lesions 1,3 and demyelination in rodents after intracerebral injection of JHM corona virus. 28 While feline corona virus (FeCV and the mutated form FIPV) infection does occur in the captive cheetah population, 9,12 FIPV causes extensive inflammation of the meninges, choroid plexus, and ependymal lining of the ventricles in felids and does not cause a viral-induced leukoencephalopathy.
While the lesions and clinical picture of LFL are inconsistent with any previously described viral disorder, early investigations included immunohistochemical screening for several viruses, including coronvirus, canine distemper virus, and polyoma virus. No FIPV or canine distemper virus antigens were identified in immunohistochemical-stained sections from 20 cases reviewed early in the epidemic, nor were polyoma virus antigens detected in an additional 3 cases screened by immunohistochemistry for polyoma virus T antigen (data not shown). LFL cases screened by electron microscopy for virus particles, while including some astrocytes and gitter cells with intracytoplasmic 70- to 90-nm spherical structures (data not shown), did not have particles with discrete membranes and internal structure typical of most viruses.
LFL appears to be a new disease. While cheetahs have been extensively studied since 1988, the first case in this species did not appear until 1996, and the first documented case of LFL was diagnosed in a Florida panther in 1994. The disorder either did not exist or escaped detection in large felids before 1994. The overwhelming predominance in cheetahs may be due to a species predilection or may be boosted by sampling bias, as this species is managed by a comprehensive central monitoring program. Once identified, the disease was closely monitored in the captive cheetah population from which it disappeared in 2005 and, to our knowledge, has not been diagnosed since. The single case originating outside the United States was diagnosed in a captive cheetah housed in the United Kingdom. The diagnosis was made on postmortem examination of the brain following euthanasia due to neurologic disease that progressed over an 8-month period during the peak incidence of disease (1997–1998) in the United States. This cheetah was captured in Namibia and exported to the United Kingdom, where it remained at a single facility for 11 years. South African and European captive cheetahs are also systematically examined as part of a global cheetah pathology surveillance program, so it is unlikely that cases of LFL went undetected in this species outside the United States (N. Kriek, E. Lane, and N. Roberts, unpublished data). The geographic restriction of LFL almost entirely to the United States could be due to regional management practices (including food sources), although it is interesting that a myelopathy occurred in European and South African cheetah cubs at approximately the same time. 34 LFL has not been described in wild felids.
While cheetahs are believed to be the only species affected by LFL at most sites where other felid species were present and managed similarly, the fact that LFL was the primary disease in only 50% of the 70 affected cheetahs examined and that 3 individuals from the other 2 subfamilies of Felidae were also affected in the same period raises concerns that other cases in less closely monitored felids were missed.
The temporal and geographic features of LFL are supportive of an exogenous focal exposure to an agent associated with environmental or husbandry practices rather than heritable or infectious causes. Since cheetahs in the United States have common founders with European and South African cheetah populations and all populations are genetically depauperate, 23 the geographic isolation of all but 1 case to the United States cannot be explained on a genetic basis. Additionally, pedigree analysis suggested that the disease had a temporal distribution within the population rather than being specific to any lineages. It is possible, however, that the homogeneous genetic composition of cheetahs predisposed them to the disease, making them more susceptible than other large felid species. Genetic makeup could determine xenobiotic metabolizing pathways, 17 unique metabolic or physiologic responses to an insult, or atypical responses to novel pathogens. Cheetahs are homogeneic for MHC genes and other loci 22,23,35 and theoretically could respond in an unusual and stereotypic manner to an exogenous agent.
While an environmental toxin (eg, in the air or water supply) is not probable because of the widespread distribution of cases throughout the United States, exposure of captive US felids to a common neurotoxin—via diet, pharmaceuticals, or vaccines—was quite possible over the time course that LFL was diagnosed. Potential exposure should be viewed in the context of intensive health management of captive populations, particularly cheetah. Because of a high incidence of disease, US cheetahs have been more intensively managed than other felids and were more often medicated and anesthetized than either European or South African cheetah populations. Furthermore, health management became more standardized across the US population from 1988, when the Cheetah SSP Research Council recommended a research approach to population health and breeding problems. Adjustments in protocols were likely made by multiple institutions in an attempt to follow these recommendations, creating an environment in which US cheetahs were more likely to have been exposed to similar vaccines, anesthetics, and treatment regimes over the 11- to 12-year period in which LFL took place. The affected animals were among this research population and would have had the longest exposure to exogenous agents. The intensive management recommended for Cheetah SSP animals included annual examinations under anesthesia—annually and before animals were moved among facilities for breeding. Tiletamine–zolazepam–medetomidine anesthesia 7 was observed to induce hypertension in some animals, which can disrupt the blood-brain barrier, 14 potentially increasing exposure to neurotoxins. While these anesthetics were in widespread use during the study period, they were used within affected and unaffected cheetahs (data not shown); thus, data would need to be carefully scrutinized before links to a specific anesthetic drug could be associated with LFL cases. Some forms of neural hypoxia are known to produce leukoencephalomalacia, including that occurring in utero 31 and that associated with carbon monoxide poisoning. 11,30 Mineralization of astroglia, noted in some LFL lesions, may be seen with hypoxic damage, and lesions similar in character and distribution to cheetah LFL have been induced in domestic cats by carbon monoxide exposure. 13 As with anesthetic exposure, vaccine use in US captive felid populations is an avenue for epidemiologic investigation. Vaccine use may have increased during the period of LFL diagnoses, and consequently affected animals in the United States could have received more vaccines over their lifetime than previous US animals did and more than non-US animals did in countries where LFL has not be noted. Aluminum, which was present in many vaccine adjuvants at this time, is a potential neurotoxin that is taken up by astrocytes more than neurons 4 and could impair normal function. Aluminum also increases vascular permeability and is associated with abnormal myelination of the spinal cord. 10 Furthermore, blood aluminum increases with renal dysfunction, a common concurrent disease in animals with LFL.
The majority of neurotoxic exposures in humans occur through food, 17 and intoxications in animals and humans through point source distribution are well documented. Precedent for a relationship between food source intoxication and development of white matter degeneration can be found in the leukoencephalopathy, primarily of the spinal cord, of domestic felids fed irradiated dry-food diets. 5,8 The mechanism of this lesion, currently unknown, may reflect a uniquely feline susceptibility to some toxins or diet-associated reactive O2 species capable of activating a cascade of mediators that can cause CNS lipid peroxidation. During the period of LFL, the majority of captive cheetah populations were concentrated in a relatively small number of institutions, several of which used the same vendor for their large felid diets. A detailed examination of vendors used and methods of packaging, processing, and distributing meat products to these institutions during the 1990s may help to resolve lingering questions about a diet-related toxic exposure.
The youngest animal diagnosed with LFL was 7 years old, and the disease was most prevalent in the aged population. Aging, in addition to potentially allowing longer exposure to neurotoxicants, likely contributes to the pathogenesis of LFL more generally. Cerebral white matter astroglia increase with age in many species, 16,27 while the ability of the CNS to limit oxidative damage through enzymatic degradation, 2 the metabolism of drugs, and immune function all decline with age. 17 Together, these features are thought to predispose the aging brain to a number of neurodegenerative conditions. Detailed serial imaging studies would have been necessary to accurately document the onset and progression of LFL lesions and possible resolution of lesions with removal of the inciting cause; thus, there remains a possibility that onset may have occurred at younger ages than what postmortem data could identify. It is also not possible to rule out resolution of preclinical neurologic changes.
LFL had a catastrophic effect on the US cheetah population through loss of valuable breeding animals, and it remains unclear how many other captive felids may have experienced the disease. Because comprehensive pathology programs that include histopathologic examination of the brain existed at a number of institutions across the United States in the 1990s, it is not believed that the lesion of LFL was missed in great numbers. However, it may prove very important for investigators pursuing cause and pathogenesis to understand that LFL appears to have first emerged in a panther and that large felids in addition to cheetahs are susceptible. The fact that LFL has apparently disappeared without identification of its cause leaves large felid populations vulnerable to its return and puts the onus of diligent neurologic surveillance on the facilities that house them and the veterinary pathology community that monitors them. It is our hope that mining the epidemiologic data from the period in which LFL occurred will eventually elucidate the cause of this disease.
Footnotes
Acknowledgments
We would like to thank the following people for their consultation over the greater-than-10-year period in which this study was conducted: Diane Nayden, Bob Nordhaussen, Teena Smith, Aimee Norris, Bob Higgins, Tony Palmer, James Powers, Albee Messing, Barbara Crain, Bill Blakemore, Mari Golub, James Evermann, Fred Murphy, Bruce Madewell, Beth Hammond, and John Lewis. We also thank the veterinary, keeper, and animal manager teams of participating zoological institutions that made early behavioral observations of affected cheetahs and provided invaluable diagnostic materials in partnership with the Association of Zoos and Aquariums’ Cheetah Species Survival Plan.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Financial support for this research was provided in part from the Association of Zoos and Aquariums’ Cheetah Species Survival Plan.