
Abstract
Once focused mainly on the characterization of neoplasms, immunohistochemistry (IHC) today is used in the investigation of a broad range of disease processes with applications in diagnosis, prognostication, therapeutic decisions to tailor treatment to an individual patient, and investigations into the pathogenesis of disease. This review addresses the technical aspects of immunohistochemistry (and, to a lesser extent, immunocytochemistry) with attention to the antigen-antibody reaction, optimal fixation techniques, tissue processing considerations, antigen retrieval methods, detection systems, selection and use of an autostainer, standardization and validation of IHC tests, preparation of proper tissue and reagent controls, tissue microarrays and other high-throughput systems, quality assurance/quality control measures, interpretation of the IHC reaction, and reporting of results. It is now more important than ever, with these sophisticated applications, to standardize the entire IHC process from tissue collection through interpretation and reporting to minimize variability among laboratories and to facilitate quantification and interlaboratory comparison of IHC results.
Keywords
Paul Ehrlich introduced the term antibody (Antikörper) in 1891 46 and hypothesized that cell surface receptors bind specifically to toxins in a lock-and-key interaction. However, it was not until 1941 that antigen detection in tissue sections was reported, marking the birth of immunohistochemistry (IHC). 65 Since then, the number of tests and their specificity and sensitivity have increased to the point that IHC routinely supplements the morphologic approach to pathology. 292 Although a major early application was in the characterization of neoplasms, IHC currently has broader and more clinically oriented applications in diagnosis, prognosis, therapeutic decision making, and pathogenesis. 206,372 In essence, IHC fills the gap between classic histopathology and molecular pathology.
Immunohistochemistry bridges 3 disciplines: immunology, histology, and chemistry. The fundamental concept is the demonstration of antigens within tissue sections by means of specific antibodies. The immunoglobulin molecule has binding sites for antigens and for other antibodies (Fig. 1). Antigen-antibody binding is demonstrated with a colored histochemical reaction visible by light or fluorescent microscopy. 284 In other words, the basic principle of IHC, as with other “special” histochemical methods, is visual localization of cell or tissue target molecules based on a satisfactory signal-to-noise ratio. 371 Although conceptually simple, the methodology of IHC has become more complex with more stringent requirements for sensitivity and specificity. 237 The initial simple (direct) methods produced quick results but lacked sensitivity. Currently, extremely sensitive methods can detect 1 or multiple antigens simultaneously or can screen hundreds of tissues in the same section (tissue microarray technology) for the presence of a particular antigen. The feasibility of detecting multiple antigens by IHC has improved dramatically with the use of multispectral analysis. 209 Another critical advance in the 1990s was the introduction of techniques to “retrieve” antigens that had been altered by fixation, increasing exponentially the number of antigens detectable in routinely fixed tissues. 284
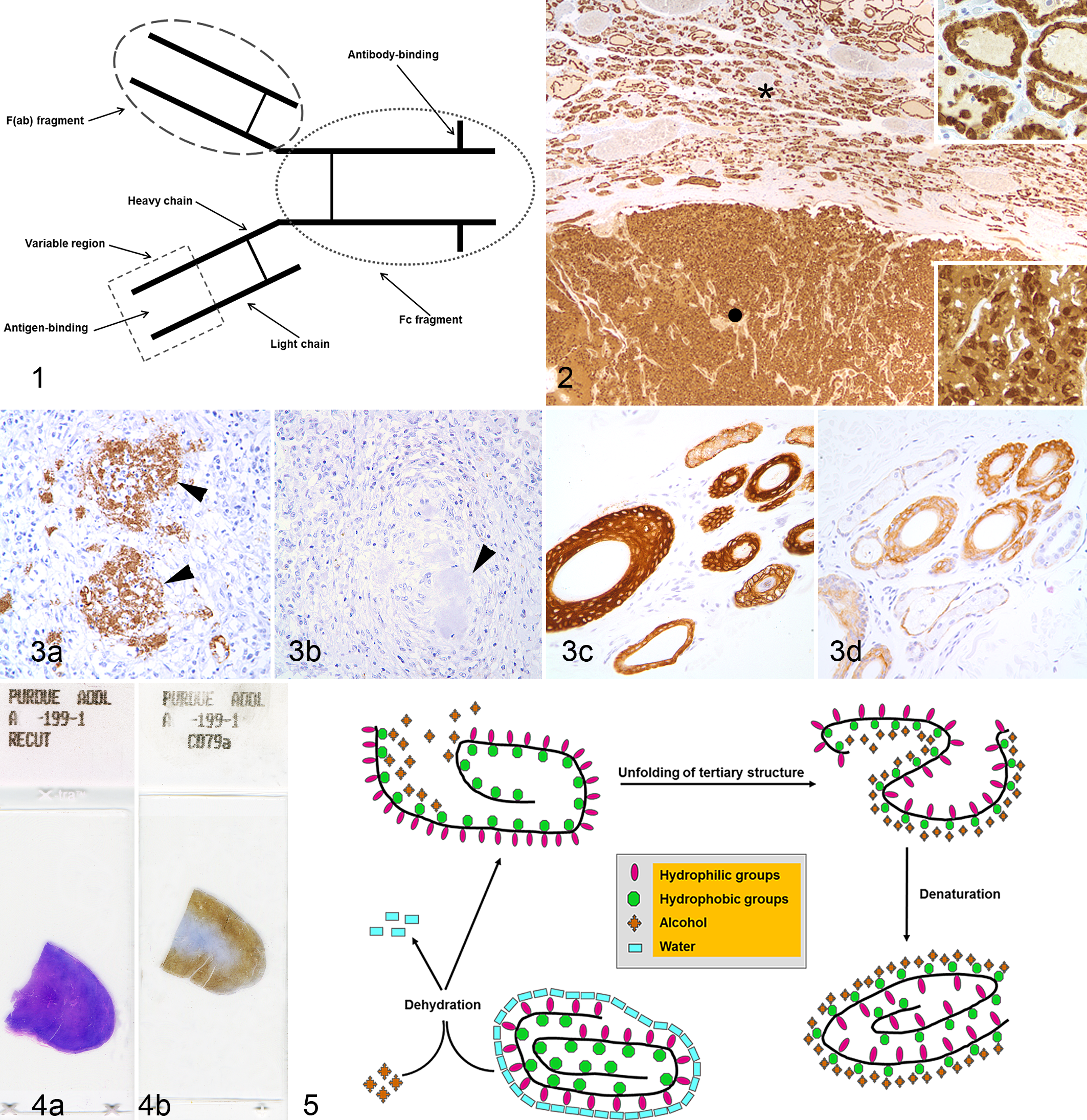
Immunoglobulin structure. The Fc fragment is delimited by the oval dotted line; the F(ab) fragment, by the oval dashed line. The area within the square is the variable region of the F(ab) fragment.
In some cases (eg, prion diseases), IHC is considered the gold standard to which other techniques are compared. In contrast to many detection techniques, IHC allows colocalization of an antigen with a lesion, thereby dramatically enhancing diagnostic interpretation and understanding of the pathogenesis.
Although IHC has become a routine tool for veterinary diagnostic and research studies, the variable cross-reactivity of antibodies among different species raises many challenges for the comparative pathologist. 284 This review addresses technical aspects of IHC—fixation, antigen retrieval, antigen-antibody reactions, detection methods, controls, standardization, validation, and quality assurance—as well as interpretation. Basic aspects of immunocytochemistry will be addressed briefly.
Overview of the Immunohistochemical Test
The IHC technique is a combination of immunologic and chemical reactions visualized with a photonic microscope. The technique can be divided into 3 phases (Table 1). Phase 1 (preanalytical) starts with sample procurement, followed by tissue fixation, trimming, and embedding, and ending with tissue sectioning on a microtome. Phase 2 (analytical) starts with deparaffination of tissue sections; includes preincubation steps (eg, antigen retrieval, blocking of nonspecific activities), incubation with the primary antibody, and labeling of the antigen-antibody reaction; and ends with slide counterstaining and coverslipping. Phase 3 (postanalytical) includes interpretation of results and generation of an IHC report, as well as evaluation of the IHC controls. 369
Steps and Variables in an Immunohistochemical Test.
HIER, heat-induced antigen retrieval; IHC, immunohistocehmistry.
Preanalytical Phase of IHC
Fixation
Fixation of tissues is necessary to (1) adequately preserve cellular components, including soluble and structural proteins; (2) prevent autolysis and displacement of cell constituents, including antigens and enzymes; (3) stabilize cellular materials against deleterious effects of subsequent procedures; and (4) facilitate conventional staining and immunostaining. 142 No fixative optimally fulfills all these goals. 284 Chemical fixation is generally used for diagnostic IHC; the most common chemical fixative is formalin (Table 2).
Comparison of Fixatives Used in Immunohistochemistry.
Modified and used with permission by the Clinical and Laboratory Standards Institute, document I/LA28-A2. 149
aFixatives containing mercury require special handling and disposal procedures.
Effects of Delayed Fixation
Prompt transfer of postmortem specimens into fixative is critical. 149 Delayed fixation can decrease the mitotic index, with up to 40% reduction when fixation is delayed more than 12 hours; 70,82,151 this reduction in mitotic index can alter tumor grade. 356 Likewise, with biopsy specimens, once the tissue is removed from the living body, biochemical alterations include adenosine triphosphate (ATP) depletion and disruption of sodium, potassium, and calcium gradients. 149 Prolonged hypoxia as a result of delayed fixation can cause cellular swelling, generation of reactive oxygen species, and activation of various enzymes, all of which can change the immunoreactivity of the target antigen, especially for measurement of apoptosis or detection of the phosphorylation state of signaling pathways. 94,149 Delayed fixation can affect the IHC results in various ways. 79,94,149,223,272 A fixation delay of more than 2 hours can decrease the detection of breast cancer biomarkers such as estrogen receptor (ER), progesterone receptor (PR), and HER2, 186 although others have not observed differences in ER and PR reactivity in samples stored in similar conditions (4°C) for several days. 5 Enzyme-rich tissues, such as intestine or pancreas, autolyze rapidly; proteolysis can lead to increased background staining (Fig. 2). 149 Diffusion of soluble proteins with fixation delay has been reported with thyroglobulin, myoglobin, glial fibrillary acidic protein (GFAP), and other cellular proteins. 95,184,355 Protozoa and fungi may be more resistant to autolysis than viruses. 291 If delay in fixation is anticipated, samples should be refrigerated. 149
The quality of fixation is influenced by the type of fixative; fixative pH, buffers, concentration, osmolality, and additives; fixation time and temperature; and the use of postfixation procedures (eg, decalcification). Two types of fixatives are used in histopathology: cross-linking (noncoagulating) and coagulating. The choice of fixative determines the need for pretreatments (eg, antigen retrieval), titer of the primary antibody, the intensity of the specific reaction versus background (signal-to-noise ratio), and even the detection pattern of antigens. 149,264
Formaldehyde: A Cross-Linking Fixative
Formaldehyde is the standard fixative for routine histology and IHC. Two factors contribute to the use of formaldehyde as the fixative of choice for most histologic procedures: (1) for more than a century, pathologists have studied tissues fixed in formalin and have become accustomed to histologic examination through the artifacts it produces, and (2) archived paraffin blocks, an invaluable resource for disease studies, most commonly contain formalin-fixed tissues. 85 Formaldehyde preserves mainly peptides and the general structure of cellular organelles. It also interacts with nucleic acids but has little effect on carbohydrates. 91,194 It is a good preservative of lipids if the fixative contains calcium. 176
The Chemistry of Formalin Fixation
Fixation with formaldehyde is a 3-step process of penetration, covalent bonding, and cross-linking. 51,77 Formation of cross-links between target peptides and irrelevant proteins reduces or blocks their immunoreactivity. 39 Whereas these steps happen simultaneously, they do so at different rates, with penetration being about 12 times faster than bonding and the latter 4 times faster than cross-linking. 51 For example, a 3-mm-thick sample will be 100% penetrated, 24% bonded, and 6% cross-linked in 8 hours; after 24 hours of fixation, it will be 70% bonded and 36% cross-linked. 53 Fixation at 37°C is faster than at 25°C. 145 Notably, formaldehyde penetrates tissues rather quickly, but tissue penetration does not equal tissue fixation. Tissue fixation by formaldehyde is considered a clock reaction because formaldehyde is in equilibrium with methylene glycol. 149 Only formaldehyde (and not methylene glycol) produces the characteristic cross-linking formaldehyde fixation. 109,149 As formaldehyde is used during tissue fixation, methylene glycol is converted into formaldehyde to maintain the equilibrium; this newly formed formaldehyde reacts with proteins, producing additional cross-links and therefore further fixation. 149 In solution, formaldehyde binds the following amino acids: lysine, tyrosine, asparagine, tryptophan, histidine, arginine, cysteine, and glutamine. 235,261,334 Therefore, knowing the amino acid composition of an epitope may predict its sensitivity to fixation in formalin. 348 However, the cross-linking between antigens and unrelated tissue proteins also has a marked effect on the immunoreactivity of antigens after fixation. 347 Cross-linking due to aldehyde fixation can produce new epitopes that specifically react with antibodies designed for another purpose (eg, the specific detection of enamel proteins in glutaraldehyde-fixed tissue by an antibody to vimentin). 178
The basic mechanism of fixation with formaldehyde is the formation of addition products (adducts) between the formalin and uncharged reactive amino groups (-NH or NH2) that eventually will form cross-links. 75 The formation of methylol adducts inactivates nucleases and proteases. 261 Once the addition product (reactive hydroxymethyl compound) is formed, additional cross-links develop. Thus, in the presence of a second reactive hydrogen, the hydroxymethyl group forms a methylene bridge.
1. Formation of addition products
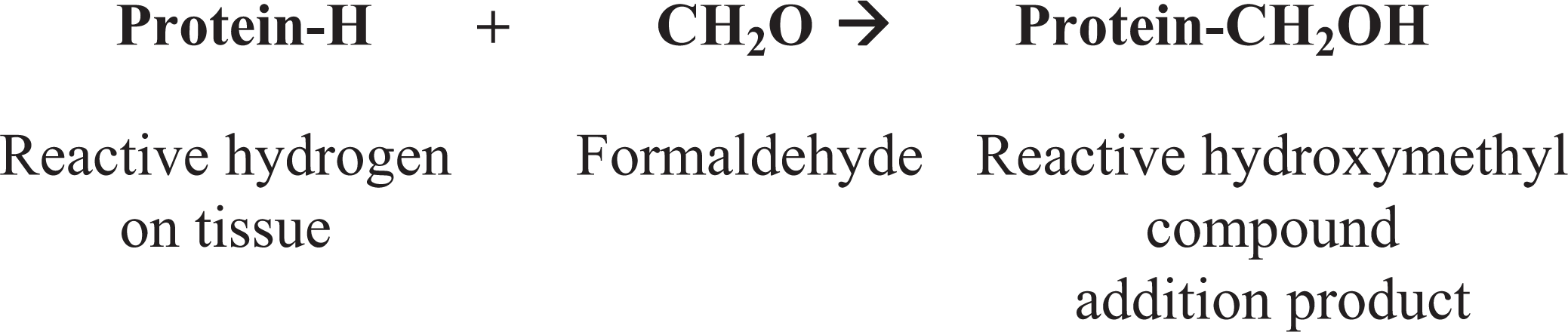
2. Formation of methylene bridges
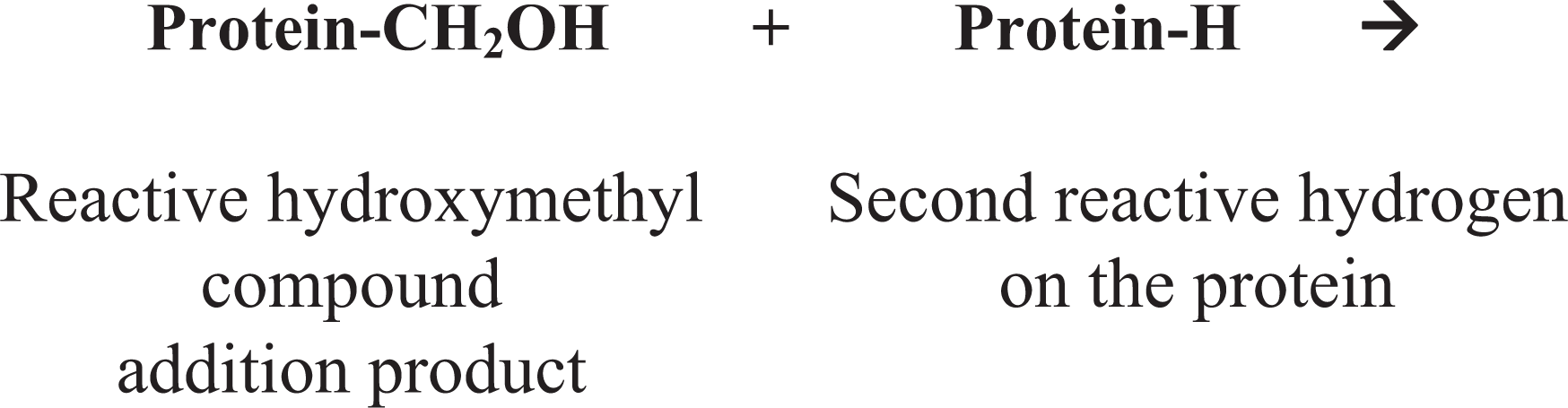

The final result of formaldehyde fixation is a profound change in the conformation of macromolecules, which may make the recognition of proteins (antigens) by antibodies difficult or impossible. 245 Although the primary and secondary structures of proteins are relatively spared, formalin fixation alters the 3-dimensional (tertiary and quaternary) structure of proteins. 142,231 Changes in the tertiary structure may not be the direct result of formaldehyde fixation but rather the subsequent interaction of cross-linked proteins with ethanol or clearing agents during tissue processing. 77,108,261 More energy (eg, antigen retrieval) is needed to reverse formalin fixation cross-links after processing tissues in ethanol, confirming that antigen immunoreactivity is affected not only by formalin fixation but also by postfixation procedures. 108
The deleterious effects of formaldehyde on immunoreactivity in cells and tissues can be partially reversed, 92 whereas glutaraldehyde fixation is considered irreversible. 91 Prolonged washing of fixed tissues reduces further fixation by removing unbound formaldehyde, 142 although established cross-links remain. 92 Long-term storage of formalin-fixed tissues in alcohol halts the formation of cross-links and, therefore, facilitates antigen detection should the tissues be needed subsequently for IHC. Overfixation can also be partially corrected by soaking tissues in concentrated ammonia plus 20% chloral hydrate. 210
The pH of a fixative buffer dramatically influences the degree of cross-linking. 92,113,282 Amino acids are charged (-NH3 +) at lower pH and uncharged (-NH2) at higher pH. When using neutral buffered formalin, the pH is shifted to neutrality, causing dissociation of hydrogen ions from the charged amino groups (-NH3 +) of the protein side chains, resulting in uncharged amino groups (-NH2). These uncharged groups contain hydrogen ions that can react with formalin to form addition groups and cross-links. In other words, the use of 10% buffered formalin (pH 6.8–7.2) produces more cross-links than nonbuffered formalin and therefore more deleterious effects for IHC. 284 If the fixative is acidic formalin, cross-linking is reduced, so the antigen retrieval procedure may need modification. In comparison, 10% neutral buffered formalin, 10% formal saline, and 10% zinc formalin were each superior to 10% formal acetic in preserving antigenicity. 412
After an initial drop in immunoreactivity with fixation, there is a plateau of variable duration before antigens become unrecognizable by specific antibodies. 347 Overfixation could produce false-negative results due to excessive cross-links, 142 especially before the advent of heat-based antigen retrieval methods. 205 However, significant reduction of immunoreactivity is detected in only a few markers after several weeks’ fixation (Fig. 3), 238,403,404 so it seems that the negative effects of overfixation are antigen and antibody dependent and can be overcome in many instances with an appropriate antigen retrieval procedure. 6,22,164,242 Vimentin was once used as an internal control of antigenicity loss in overfixed tissues. 18,306 However, with heat-based antigen retrieval, some antigens in overfixed tissues may not be well preserved even if vimentin reactivity does not indicate antigen degradation. 6 The effect of prolonged formaldehyde fixation on an antigen depends on its cellular localization 142 —for example, there is irreversible loss of Bcl-2 immunoreactivity in the nucleus, even with heat-based antigen retrieval, whereas cytoplasmic immunoreactivity is preserved or increased after antigen retrieval. 154
Underfixation can also produce unexpected results and is considered a more common and serious problem than overfixation. 35,149 In a typical diagnostic setting, formalin-fixed tissues are processed through a series of alcohol gradients prior to paraffin embedding. If large samples are fixed only 24 to 48 hours or small biopsy specimens for only several hours, cross-links may develop only in the periphery of the specimen with the core of the tissue left unfixed or fixed only by coagulation with the alcohol used for dehydration during tissue processing, 142,405 resulting in a IHC gradient across the section (Fig. 4). 149 Underfixation can produce irrelevant or equivocal results that can alter the detection or scoring of some biomarkers. 123,171 Antigen retrieval of underfixed tissues may result in unexpected antigen detection. 284 Underfixed tissues are easily damaged by harsh antigen retrieval methods, with subsequent loss of antigenicity. 303
There is no optimal fixation time for every antigen. The structure of the antigen as well as its relationship with other proteins probably influences the effect of fixation on immunoreactivity. The current Clinical and Laboratory Standards Institute recommendation for formalin fixation of diagnostic specimens is 16 to 32 hours; on average, complete fixation is achieved after 24 to 48 hours. 149 Interestingly, the recommended 10:1 ratio of fixative to sample was challenged when a 2:1 ratio and 48-hour fixation at room temperature was considered sufficient for routine histology and IHC. 53 Fixative penetration, bonding, and cross-linking are faster at higher temperatures. Samples to be fixed should not be thicker than 2 to 4 mm. Although formaldehyde has been deemed a suboptimal fixative for IHC, with optimal antigen retrieval procedures, it is satisfactory for most antigens.
Glyoxal, the Ideal Formalin Substitute?
Glyoxal is a dialdehyde that resembles 2 back-to-back formaldehyde molecules. 76 Whereas formaldehyde forms a single hydrated species (methylene glycol), glyoxal has several hydrated forms, the most common of which is 1,3-dioxolane. 76 One advantage of glyoxal over formaldehyde is that it does not emit vapors. In addition, glyoxal has poor reactivity toward most end groups and forms addition compounds only with arginine, lysine, cysteine, and the single α-amine group of proteins, whereas formaldehyde reacts at room temperature (RT) with almost all end groups that contain nitrogen or oxygen to form single-carbon adducts. 76 In comparison to formaldehyde fixation, glyoxal fixation is faster with minimal or no cross-linking, rendering antigen retrieval unnecessary for many antigens. Although a special antigen retrieval solution with high pH can be used, standard antigen retrieval procedures in glyoxal-fixed tissues may be ineffective or even deleterious. 76 The perceived advantages of glyoxal were disputed, however, in a quantitative image analysis study in which formaldehyde was considered superior in terms of morphologic preservation and IHC signal. 59
Coagulative Fixatives
The problems with formaldehyde fixation, especially the loss of immunoreactivity (particularly before the development of heat-based antigen retrieval methods), have prompted a search for alternative fixatives. 284 Many formalin substitutes are coagulating fixatives that precipitate proteins by breaking hydrogen bonds without protein cross-linking. The most common types of coagulative fixatives are dehydrants (alcohols and acetone) and strong acids (picric acid, trichloroacetic acid). Most body fluid proteins have hydrophilic moieties in contact with water and hydrophobic moieties in closer contact with each other, stabilizing hydrophobic bonding. Removal of water by ethanol destabilizes protein hydrophobic bonding because the hydrophobic areas are released from the repulsion of water, and the protein tertiary structure unfolds (Fig. 5). 92 Simultaneously, removal of water destabilizes hydrogen bonding in hydrophilic areas. The resulting protein denaturation causes inadequate cellular preservation 142,153,401 and a possible shift in intracellular immunoreactivity as reported for some growth factor peptides and cytokines (Fig. 6). 42,383 Coagulative fixation maintains tissue structure at the light microscopic level fairly well but results in cytoplasmic flocculation as well as poor preservation of mitochondria and secretory granules. 284
The Search for Alternatives to Formaldehyde Fixation
There is no “one fixative fits all” for IHC. 128 Formaldehyde is used mainly because it is reliable for general histology, it is inexpensive, and its deleterious effects can often be countered with antigen retrieval. 280 In addition, the interpretation of retrospective studies would be onerous if archived paraffin blocks contained tissues preserved in a variety of fixatives over the years. The value of nonformaldehyde fixatives for research purposes has been demonstrated for various antigens.* Most available formalin substitutes are alcohol solutions; therefore, fixation is based on dehydration and protein coagulation. 51,83,198 Weigners fixative, a mixture of alcohols and pickling salt, appears to be superior to formaldehyde for DNA and RNA studies, performed comparably to formaldehyde for IHC, and maintained immunoreactivity for some biomarkers after prolonged fixation when formaldehyde did not. 193 Carnoy’s fixative, a mixture of alcohol–chloroform–acetic acid, was superior to formalin for IHC detection of some antigens without antigen retrieval in tissue-engineered constructs. 195 Tissues fixed in PAXgene, which consists of a fixation reagent (methanol, acetic acid, and a organic solvent) and a postfixation stabilization reagent (alcohol mixture), may not need antigen retrieval for some antigens, 183 although immunoreactivity for other antigens is reduced compared with formalin-fixed, paraffin-embedded (FFPE) tissues. 21 For molecular analysis, PAXgene preserves RNA better than does formalin. 130
The immunoreactivity of antigens in fixed and paraffin-embedded tissue varies markedly depending on the antigen and more specifically on the epitope recognized by the antibody. 8,59 In a comparison of the effect of different fixatives (strong cross-linkers, weak cross-linkers, coagulant, and combination coagulant/cross-linkers) on antigen immunoreactivity in mouse tissue, formaldehyde, followed by the combination fixatives, performed better than the other types. 59 Substitution of a different fixative in an IHC test validated for formalin-fixed tissue demands a new validation study. 149
Microwave Fixation
The use of a microwave can reduce fixation time and therefore the overall time for tissue processing. After immersing the tissue in formalin at RT for at least 4 hours, adequate fixation can be achieved in a 5-mm-thick sample by microwaving in formalin solution for 1.5 to 4 minutes at 55°C. 149 Validation of microwave fixation procedures for IHC mandates the use of tissue controls processed in a similar way. Microwave procedures require approved laboratory microwaves and, because of the release of formalin vapors, must be performed in a fume hood.
Decalcification
Decalcification with weak acids does not seem to interfere significantly with IHC for most antigens, provided the tissues were well fixed in formalin; 284 however, decalcification with strong acid solutions has a negative effect on immunoreactivity, at least for some antigens. 7,9,96,232 In a study of melanocytic markers in canine tissues, decalcification for less than a week with formic acid did not significantly reduce immunoreactivity for Melan A, PNL2, or tyrosinase, whereas the use of HCl in the decalcifying solution reduced the immunoreactivity for Melan A and PNL2 after only 1 day of treatment with complete loss of immunoreactivity after 1 week. 295 In that study, tyrosinase was more resistant than the other 2 markers to the deleterious effects of HCl decalcification. The antigenicity of intermediate filaments appears to be more resistant to decalcification effects than other antigens. 149 Antigen retrieval in a boric acid solution at 60°C seems to improve the immunoreactivity of some antigens in decalcified tissues. 414 In summary, weak (eg, formic) acid decalcifying solutions diluted in formalin are recommended for IHC. Due to the potential negative effects of decalcifying solutions, the IHC report should indicate the type, if any, of decalcification.
Tissue Processing and Incubation Buffers
Although fixation is paramount in the outcome of the antigen-antibody reaction, the incubation buffer and tissue-processing solutions can also alter antigenicity. 89,143 The combination of cross-linking fixatives with heat and the nonpolar solvents used in paraffin embedding is thought to modify the antigen conformation so that specific epitopes may not be recognized by antibodies that would recognize those epitopes in frozen sections. Thus, fixation may not be the sole factor in failure to detect an antigen. 89,113,129 There is also evidence for a cumulative effect of fixation and tissue-processing factors in the failure of antigen recognition in FFPE from a study of Ki67 and proliferating cell nuclear antigen (PCNA) immunoreactivity in formaldehyde-fixed tissues processed with alcohol and xylene. 77,264 Shifts in the tertiary structure of proteins so that hydrophobic areas are oriented outward and hydrophilic regions inward (the so-called hydrophobic inversion) 77 during dehydration and clearing steps of tissue processing can reduce or abolish antibody binding without antigen retrieval, especially with poorly stabilized (unfixed or suboptimally fixed in formalin) tissues/proteins exposed to a weakly polar or nonpolar solvent. 264 This negative effect varies with the dehydrating and clearing agent used. 93 There is also increased background reactivity in tissues left in xylene for prolonged periods during processing. 149
Few authors have evaluated the effects of long-term storage of formalin-fixed tissues in other solutions. 93,359,363 Formalin-fixed tissues stored in 70% ethanol for several weeks had no apparent loss of immunoreactivity or nucleic acid amplification. 359 Tissues stored in tap water for up to a week had no loss of antigenicity or changes in microscopic appearance. 363
The effects of dewaxing with organic solvents on IHC have not been studied systematically. However, nonorganic paraffin solvents (eg, dishwashing detergents) have been substituted for organic solvents with comparable results. 52,148
Tissue Microarrays
Tissue microarrays (TMAs), introduced by Kononen et al, 197 allow simultaneous examination of hundreds of tissues on a single microscope slide. A tissue core is transferred from the “donor” paraffin block to the “recipient” block, which may contain up to 1000 cores. 344 This technology is used not only for protein detection but also for gene expression. † Tissue microarray techniques include multitumor arrays (samples from tumors of multiple histological types), progression arrays (samples of normal tissue and different stages of tumor progression within a given organ), prevalence arrays (tumor samples from 1 or several entities without extensive clinicopathologic information to test biomarkers with possible therapeutic implications), prognosis/outcome-based arrays (samples with clinical follow-up data to evaluate prognostic or predictive biomarkers), cell line arrays (normal or cancer cell lines grown in culture to determine the specificity of antibodies targeting a protein), heterogeneity/random tissue/tumor arrays (tumor and nontumor tissues from different sites for monitoring the intratumoral heterogeneity of molecular markers), and cryomicroarrays (frozen samples that might be more suitable than formalin-fixed tissues for RNA detection). 256,316,324,344,367
The advantages of the TMA method include less reagent consumption; decreased technical time; decreased variability of results; the possibility of digitizing and quantifying results or interpretation of results by hierarchical cluster analysis, quality control, and standardization; evaluation of antibody sensitivity and specificity; and rapid and high-throughput discovery and validation of biomarkers. 173,258,305,311,358 With an adequate selection of control tissues, fewer than 12 tissue cores in an array suffice to evaluate more than 90% of the markers used in diagnostic IHC. 150 With tissue microarray, the cell expressing a particular gene can be identified, whereas in DNA microarray, the sample is digested before testing, so cell expression cannot be localized. Although the concept of tissue microarray is simple, this method has some disadvantages compared with the classic single sample/microscope slide: preparation (with a commercial manual or automated array) requires high technical skills and careful planning, sample selection is critical due to its small size and the usual heterogeneity of the sample, and differences in fixation protocols in archival tissue can influence TMA results. 99,172,173,266,358
Sampling of multiple and different areas of the donor block using small (0.6-mm) cores can be more representative of the lesion and less likely to damage the block than extracting a single and larger core. 99,324 Results from triplicate TMA cores had up to 98% concordance with the results from full-block sections, whereas concordance was lower with only 1 or 2 cores. 99,119 Nevertheless, intratumor heterogeneity can be a complicating factor when assessing biomarkers in TMAs, 212 especially when a scoring system (eg, 0, 1+, 2+, 3+ for HER2 in breast cancer) for the IHC reaction is used for therapeutic planning.
The current Clinical and Laboratory Standards Institute (CLSI) guidelines for IHC recommend TMAs for internal and external quality assurance programs. However, due to various technical issues, the use of TMA for diagnostic purposes is not recommended. 125,149 The amount (diameter and number of cores) of tissue needed to test a given biomarker varies and should be based on the type of tissue/disease process and biomarker examined. 149
Multiplex immunostaining (MI) chips can be used to examine multiple antigens in the same tissue section. MI chip technology differs from tissue microarray in that MI permits the analysis of expression of as many as 50 antigens in a single specimen, whereas the microarray technology permits the analysis of a single antigen in many specimens simultaneously. 117 The main problem in applying this technology in a clinical setting is the heterogeneity of most tumors and, therefore, the possibility of false-negative results. 117
Drying of Paraffin Sections
Paraffin sections dried overnight at temperatures of 60°C or higher may have reduced immunoreactivity for some antigens. 93,137,147,412 In another study, immunoreactivity was reduced when slides were dried at temperatures higher than 68°C for more than 16 hours, whereas drying at 58°C for 24 hours did not affect immunoreactivity. 177
Storage of Paraffin Blocks and Tissue Sections
Paraffin blocks containing formalin-fixed tissues are a powerful resource for protein and nucleic acid investigations. 192 They are the archived tissue most suitable to evaluate diseases and apply the results to targeted medicine. 354 It is the authors’ and others’ 327 opinion that paraffin blocks remain stable for years in terms of antigenicity. However, only controlled studies on the effect of nucleic acid detection in archival paraffin blocks have been published. 133 Any deleterious effects of prolonged paraffin block storage on tissue antigenicity could differ among antigens and should be considered if IHC results on old paraffin blocks are inconsistent or unexpected.
Storage of unstained paraffin control tissue sections increases efficiency but may adversely affect immunoreactivity (tissue section aging/slide oxidation/biomolecule degradation). 93,127,149 These differences are epitope specific rather than protein target specific. 149 Both light and temperature contribute to the loss of antigenicity that occurs with photo-oxidation of tissue sections. 30,304 Tissue section aging is a rather common problem with nuclear antigens (eg, Ki67, ER, p53); ‡ however, in a study of animal tissues, cytoplasmic membrane antigens were more sensitive than nuclear antigens to degradation from photo-oxidation and/or ambient temperature. 304 Immunoreactivity can also decrease when tissues are suboptimally dehydrated (retained endogenous water) during paraffin embedding or with paraffin sections stored under high humidity. 420 To reduce antigen degradation due to photo-oxidation, paraffin sections should be used within 1 week of sectioning or at least should be stored in an airtight container in the dark. 149 Keeping the paraffin sections in the dark under refrigeration also diminishes the loss of antigenicity. 304
In summary, all steps in tissue processing, from acquisition to storage, can affect the quality of the IHC test. 376 When there is decreased intensity or loss of reaction in stored control tissue sections, the IHC test should be repeated with a different stored control tissue section plus a freshly cut control tissue section. If the change is limited to stored sections, any unused stored control slides should be discarded.
Analytical Phase of IHC
Antigen Retrieval
Fixation and tissue processing modify the 3-dimensional structure of proteins, which can render antigens undetectable by specific antibodies because the immunologic reaction depends on the conformation of the antigen. 143 The purpose of antigen retrieval (AR) procedures is to reverse the changes produced by fixation. AR is particularly important for tissues fixed in cross-linking fixatives. Approximately 85% of antigens fixed in formalin require some type of AR to optimize the immunoreaction. 287 The need for AR and choice of method depend on the targeted antigen and the type of antibody; 393 AR is more commonly necessary with MAbs than with PAbs. 287 The 2 most common AR procedures in IHC are enzymatic and heat-based retrieval. Although AR facilitates detection of Ags, background staining or antigen detection in unusual locations is not uncommon with harsh AR methods and can preclude diagnostic interpretation (Fig. 7).
Antigen Retrieval With Enzymes
Protease-induced epitope retrieval (PIER), introduced in the mid-1970s, 161,162 was the most common AR method before the advent of heat-based AR in the 1990s. Commonly used enzymes include trypsin, proteinase K, pronase, ficin, and pepsin. 20,170,236,263,273 The mechanism of PIER is probably protein digestion, but this cleavage is nonspecific and some antigens may be negatively affected. 390 The effect of PIER depends on the concentration and type of enzyme, incubation parameters (time, temperature, and pH), and the duration of fixation. 20,236,273 The enzyme digestion time is proportional to the fixation time. 390 It is practical to optimize a few enzymes for laboratory use. A commercially available ready-to-use solution of proteinase K has good activity at room temperature, so can be used with automatic stainers. The disadvantages of PIER are the low number of antigens for which it is the optimal AR method and the risk of altering tissue morphology or destroying epitopes. 263,390 Interestingly, proteinase treatment has facilitated IHC detection of nucleolar proteins present in high concentration but undetectable without AR, whereas nucleolar proteins at low concentration can be detected without enzymatic treatment. 364
Heat-Induced Epitope Retrieval
Heat-induced epitope retrieval (HIER) has revolutionized the IHC detection of antigens fixed in cross-linking fixatives. In addition, HIER is used for extraction of molecules (eg, nucleic acids, proteins) from formalin-fixed, paraffin-embedded tissues. 332 HIER was introduced by Shi et al. 337 based on a concept developed by Fraenkel-Conrat et al 110–112 that the chemical reactions between proteins and formalin could be reversed (Fig. 8), at least in part, by high-temperature heating or strong alkaline hydrolysis. Although HIER denatures formalin-fixed tissues, it paradoxically restores their immunoreactivity. 348 The mechanism involves the dissociation of irrelevant proteins from target peptides. 39 The secondary and tertiary structure of proteins is probably modified (denatured) during HIER; however, that should not affect the immunoreactivity of most antigens because IHC antibodies require only an intact primary (linear) protein structure. 39,347 Likewise, some comformational epitopes become denatured after HIER and will not bind specific antibodies. 421 Heating may unmask epitopes by hydrolysis of methylene cross-links. 135,390,422,423 Rupture of cross-links resets the charged status of proteins and their hydrophobic characteristics. 421 AR also acts by other mechanisms because it enhances immunoreactivity of tissues fixed in ethanol, which does not produce cross-links. 144 Even in unfixed tissues, antigens can be unmasked by HIER, perhaps because steric barriers produced by the antigen itself had precluded antibody access to targeted intramolecular epitopes. 180
Other hypotheses to explain HIER are extraction of diffusible blocking proteins; precipitation of proteins; rehydration of the tissue section, allowing better antibody penetration; and heat mobilization of trace paraffin. 349 Tissue-bound calcium ions may mask some antigens during fixation. Calcium chelating substances (eg, EDTA) are sometimes more effective than citrate buffer in AR. 247,248,271,415 Calcium-induced changes in protein conformation may augment the immunodetection of some antigens, 399 but for many antigens, calcium-induced effects on immunoreactivity cannot be documented. 331,333 Restoring the native electrostatic charges modified during formalin fixation has also been considered an AR mechanism. 34,36
Equipment used for HIER includes decloakers (commercial pressure cookers with electronic controls for temperature and time), vegetable steamers, water baths, microwave ovens, or pressure cookers. 13,88,182,271,337 The relationship between temperature and exposure time is inverse: the higher the temperature, the shorter the time needed to achieve results. Heating at high temperature (100°C) for a short duration (10 minutes) is better than heating at a lower temperature for a longer time. 143 In most instances, the source of heat is not critical to the outcome but a matter of convenience. Satisfactory results can be obtained using a steamer (90°C–95°C) for 20 minutes for most antigens, although a decloaker eliminates the problems of irregular heating or temperature variation with a steamer or microwave oven.
A universal antigen retrieval solution is not available. 143,167 Thus, several HIER solutions made of different buffers (eg, citrate, tris, Tris-HCl) and pH (3–10) have been used. Some antigens can be retrieved with low pH solutions, others only with high pH solutions, and a third group with solutions of a wide pH range. 332,336 The importance of the chemistry of the retrieval solution, particularly the buffer, is unclear; however, for most antigens, HIER with 0.01 M sodium citrate buffer (pH 6.0) produces satisfactory results with good cell morphology when compared with buffers with higher pH or solutions containing EDTA. 19,87,143 However, use of different buffers may be required to optimize antigen recovery. A low pH buffer (acetate, pH 1.0–2.0) appears especially useful for nuclear antigens. As a rule, high pH and EDTA-containing buffers reach their optimal treatment effect faster than do lower pH citrate-containing buffers. 199 Sometimes multiple AR methods (enzyme-HIER; HIER-HIER) are needed to optimize the immunodetection of antigens (Fig. 9). 97,143,179,340,388 Triple AR was beneficial to demonstrate amyloid β. 179
The degree of fixation can dramatically modify the response of antigens to AR. Unfixed proteins are denatured at temperatures of 70°C to 90°C, whereas formaldehyde-fixed proteins are resistant at the same temperatures. 231 This could explain why the immunoreactivity of partially fixed tissue can be heterogeneous, despite the supposed even distribution of the antigen. 19 In other words, formaldehyde fixation may protect against denaturation during AR, although this has been disputed, at least when using an autoclave reaching 120°C. 39,348 For practical purposes, protein denaturation during AR does not have a negative impact on the immunoreactivity of peptides (Fig. 10).
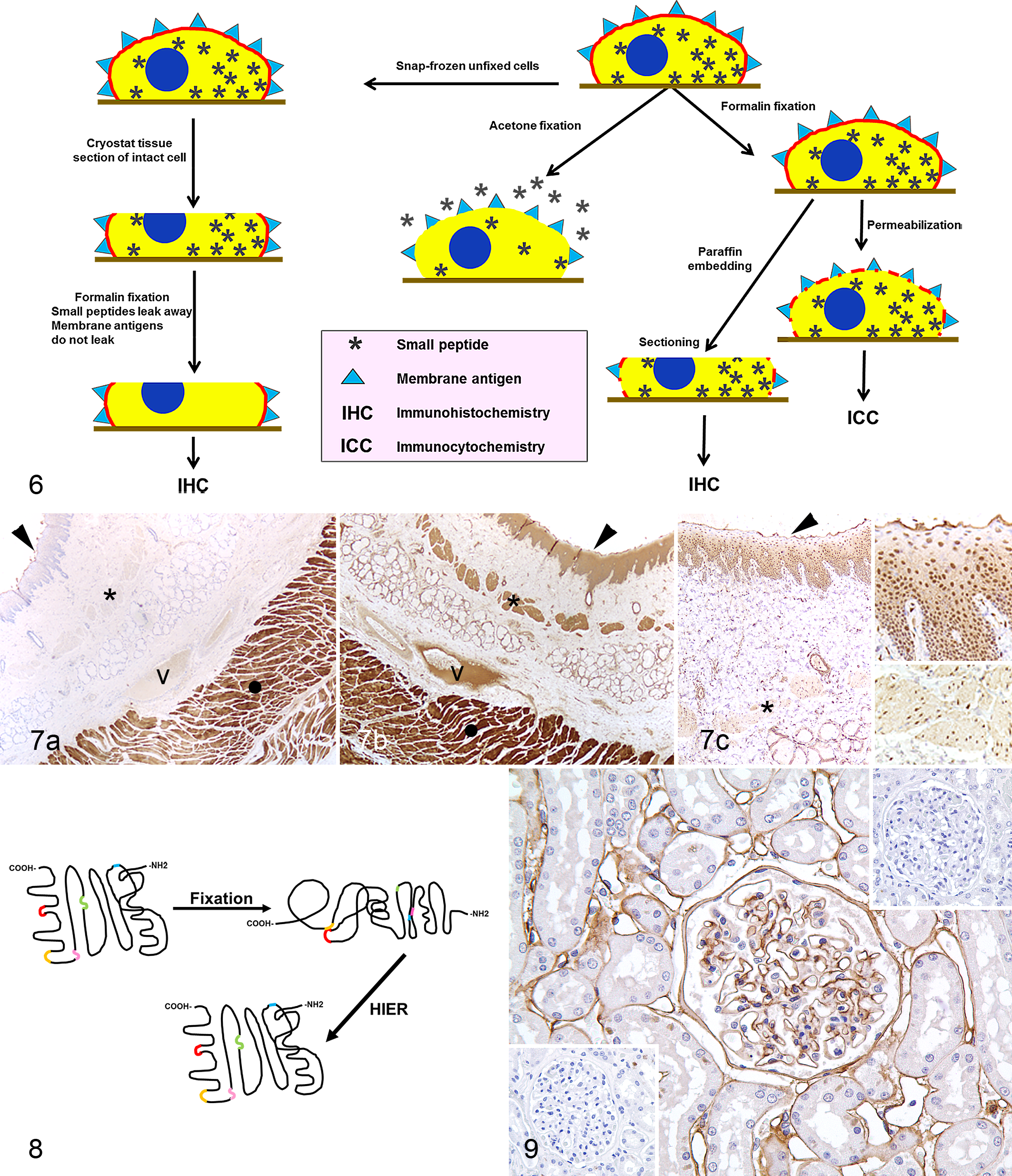
Effects of fixation on small peptides and cell membrane antigen stability. Formalin fixation stabilizes small peptides and cell membrane antigens, preventing leakage or diffusion, even after sectioning. With acetone fixation, small peptides leak through the permeabilized cell membrane. Formalin fixation stabilizes membrane antigens in cryostat sections, but small peptides leak away. Used with permission and modified from Floyd and van der Loos.
106
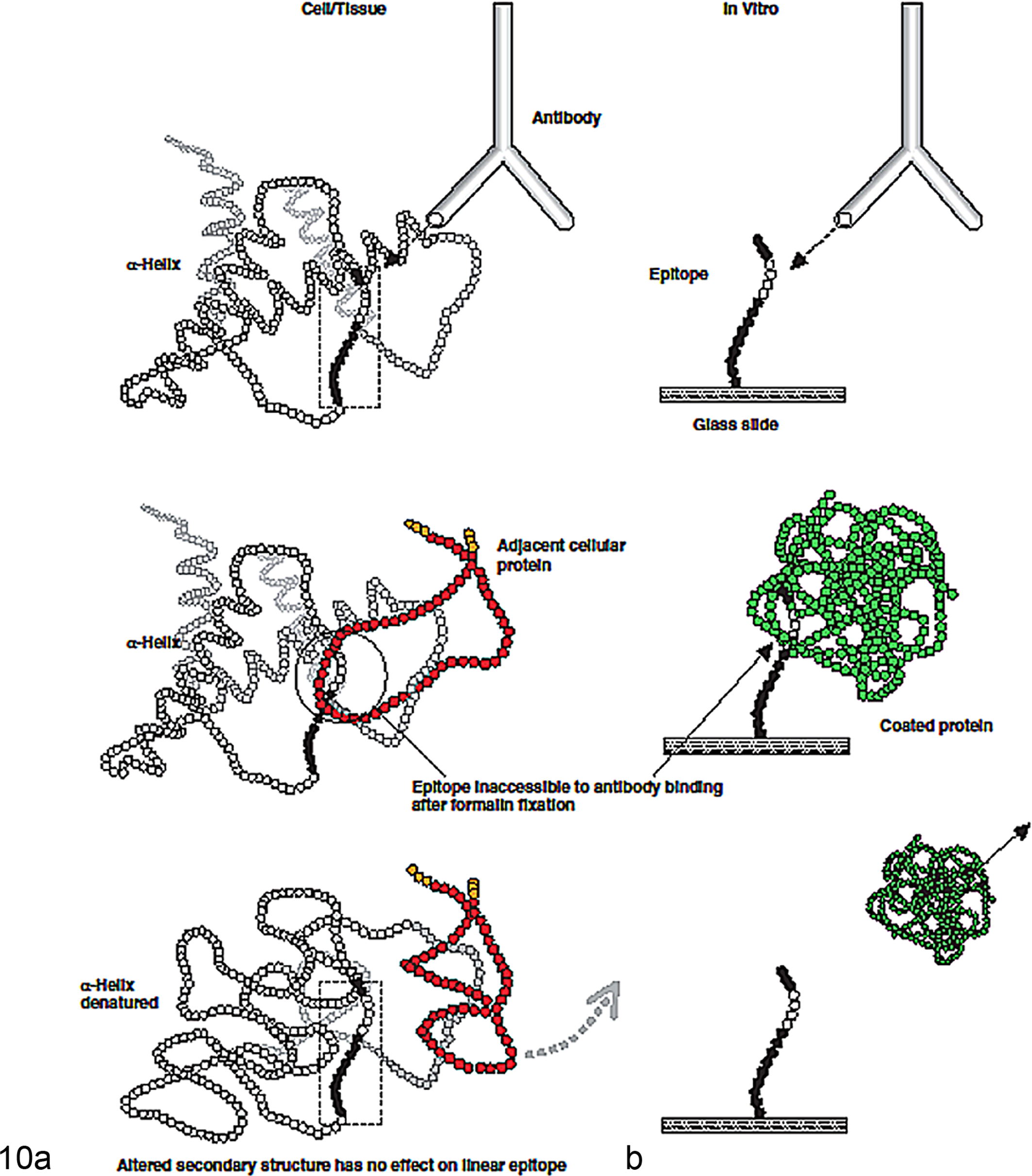
Loss of immunoreactivity after formalin fixation with recovery by antigen retrieval (AR). (a) Putative structural changes in native proteins. (b) Corresponding changes to epitopes in the in vitro peptide assay. Used with permission from Sompuran et al. 347
The variability in both the kinetics of fixation and AR for different epitopes suggests that different degrees of cross-linking may occur depending on the peptide amino acid composition. 39 The possibility of unexpected immunoreactivity should always be considered when using HIER, particularly with low pH buffers. 19 When practical, the immunoreactivity of fresh-frozen and routinely processed paraffin tissue sections should be compared 331 because not all antigens benefit from AR, even after prolonged formalin fixation. 349
Miscellaneous Antigen Retrieval Methods
Pretreatment with concentrated formic acid improves the signal in some IHC tests. 189 Other AR methods include the use of strong alkaline solution, urea, borohydride, and a solution of sucrose or acid (eg, hydrochloric acid). 330,334 Citraconic acid with heat has been used as AR to reverse formalin-induced cross-linking, presumably by hydrolysis. 249,253 This method has been successfully applied to nervous tissue fixed more than 5 years. 4 The proposed mechanism of citraconic anhydride (citraconic acid) AR is converting the cross-links in the sample to protected amines and liberating formaldehyde quickly to avoid reforming adducts; then citraconic acid liberates amines via hydrolysis. 253
Detergents, although not strictly speaking an AR component, facilitate Ag-Ab binding by solubilizing membrane proteins. 303 Detergents form mixed micelles with lipids as well as micelles that contain proteins (also known as surfactants), thereby decreasing the surface tension of water. They are usually incorporated into the dilution/rinse buffers. Examples of detergents in IHC include ionic detergents, bile acid salts, and nonionic and zwitterionic types (eg, Triton R-X100, BRIJR, Tween 20, saponin). The addition of sodium dodecyl sulfate (SDS) to the AR buffer and HIER at relatively low temperature (97°C) appears to improve the signal of many markers. 366
All-in-One Epitope Retrieval Buffers
The classic histologic procedure calls for dewaxing paraffin sections using organic solvents (eg, xylene) followed by dehydration in alcohols and hydration before AR. The so-called 3-in-1 reagents can perform aqueous deparaffination, rehydration, and AR. 149 Their use in IHC is relatively new, so comparison with traditional means of deparaffination, rehydration, and AR is required before using them in a diagnostic setting. Commercially available all-in-one epitope retrieval buffers can reduce processing time by melting the paraffin in tissue sections during HIER. 148 The advantage of these solutions is obvious, but reported instances of incomplete paraffin removal could affect the outcome of the IHC. 114 This is more commonly observed with solutions based on surfactants and wetting agents, which tend to incompletely solubilize the molten paraffin. The addition of a water-soluble organic agent appears to solve this problem without negatively affecting the IHC reaction. 114
Antigens and Antibodies: The Specificity Issue
Immunohistochemistry involves antibody recognition and binding to an epitope on the target antigen. 149 The specificity of the reaction largely depends on qualities of the primary antibody and the ability of the antigen (epitope) to bind it. The most commonly used immunoglobulin (Ig) is IgG; IgM is used less commonly. 284
Antigens
Antigens can be as small as a monosaccharide; epitopes consist of 5 or 6 amino acid residues. 149 Antigens can be multivalent with multiple identical epitopes (homopolymeric) or multiple distinct epitopes (heteropolymeric). 213 A single gene can generate several different antigen isoforms via 2 principal mechanisms. Alternative splicing of the primary gene transcript can produce multiple different mature transcripts, each of which codes for a slightly different protein. 44,328 Many proteins also undergo posttranslational modifications, such as glycosylation, phosphorylation, and proteolytic processing, which add further complexity. 237 As a result, 1 gene can generate numerous protein (antigen) isoforms, and this repertoire can change with time (eg, tenascin and hemoglobin isoforms change from fetal development to adulthood). 237
Two broad groups of immunogens are used to produce antibodies: synthetic peptides and purified protein preparations. 237 The known amino acid sequence of synthetic peptides facilitates IHC interpretation, both with respect to the isoforms of the target protein and any cross-reactivity with similar peptide sequences in other proteins. A potential disadvantage to using synthetic peptides is that an isolated synthetic peptide sequence may lack the normal 3-dimensional structure of the native protein. In addition, other proteins can be intimately associated with the protein of interest in vivo. Either factor can mask target epitopes, preventing their detection by antibodies raised to synthetic peptides and therefore yielding false-negative results. Also, crucial in vivo posttranslational modifications of the native antigen are absent in synthetic peptides. 220,221 The presence of the synthetic peptide sequence in unrelated proteins could produce immunologically specific Ag-Ab binding (molecular mimicry) despite the absence of the target antigen. 29,72,165 Use of purified proteins as immunogens avoids many of the problems of synthetic peptides. 237 However, protein purification to homogeneity from cells or tissues can be technically difficult, and contaminating proteins may be more antigenic than the protein of interest, producing a disproportionate and unwanted immunogenic response. Another problem with purified proteins arises if the targeted antigen includes highly immunogenic epitopes that are not specific to the antigen of interest. This pitfall is common when similar posttranslational modifications occur among antigens that differ markedly in amino acid sequence. Unfortunately, the source and details of antigen purification or the isoforms of the targeted protein recognized by an antibody preparation are seldom reported for commercial primary antibodies. 237 Other factors affecting immunogenicity include size, conformation, and electrical charge of the antigen; use of adjuvants; and dose and route of administration of the antigen.
Antibody Structure
Immunoglobulins are “Y” shaped and consist of 2 identical light chains and 2 identical heavy chains (Fig. 1). The heavy chains determine the antibody class. The tail of the Y, called Fc (Fragment, crystallizable), is composed of 2 heavy chains on the C-terminal side. 46 Each end of the forked portion of the Y is called the Fab (Fragment, antigen binding) region. The light chains of most vertebrate antibodies have 2 distinct forms, κ and λ. In any immunoglobulin molecule, both light chains and both heavy chains are of the same type. The light chains are divided into the C-terminal half, which is constant and called CL (Constant: Light chain), and the N-terminal half, which has abundant sequence variability and is called the VL (Variable: Light chain) region. The Fab (antigen-binding) region of the immunoglobulin has variable and constant segments of the heavy and light chains. The Fc portion determines biological functions and permits antibody binding to other antibodies, complement, and immune cells with Fc receptors. This portion of the Ig is needed for multistep IHC techniques 284 and is largely responsible for nonspecific background reactivity due to nonimmune adherence of antibodies (Abs) to the tissue section. Background can be diminished by the use of only Fab or F(ab)2 portions of the Ig molecule for IHC; however, without the Fc portion, antibody binding to solid substrates, such as tissues, is less stable. The specific binding of an antibody to an antigen occurs via hypervariable regions of both heavy and light chains of the amino terminus. 284 The antigen-binding site of an Ab is called the paratope. Epitopes, usually 5 to 21 amino acids long, are the regions of an antigen (Ag) that bind to antibodies. 141 In an immunologic reaction, one of the most important criteria for antibody binding is the tertiary structure of the epitope (ie, the way in which peptide chains of a protein are folded or interact with adjacent peptides). The paratope interacts with the tertiary structure of the epitope through a series of noncovalent bonding (see below). The more bonding, the greater the affinity and avidity of the antibody. IgG antibodies are bivalent (have 2 identical arms used in antigen recognition). This is a key component of multiple-layer immunohistochemical methods. Other immunoglobulins, except secretory IgA, which is tetravalent, and IgM, which is decavalent, are also divalent.
Epitopes are classified as linear or conformational. 39 Linear epitopes are a group of 5 to 7 contiguous amino acids. Conformational (discontinuous) epitopes, the typical form, consist of small groups of amino acids brought together by conformational folding or binding. 39 Although most epitopes involved in immune responses are believed to be conformational, data support the concept that antibodies used in FFPE tissues recognize mainly, or exclusively, linear epitopes. 39,347
The Nature of Antigen-Antibody Interactions
From a biochemical point of view, Ag-Ab interactions are somewhat unusual. 284 The bonds are weak (mostly hydrophobic, van der Waals, and electrostatic) and not covalent. Hydrophobic bonds develop between macromolecules (interatomic or intermolecular) with surface tensions lower than that of water. Hydrophobic interactions are imparted mainly through the side chain amino acids phenylalanine, tyrosine, and tryptophan. 31 By their lower attraction to water molecules, these amino acids tend to link with one another. Electrostatic (Coulombic) interactions (also called ionic bonds) are caused by attractive forces between 1 or more ionized sides of the antigen and oppositely charged ions on the antibody-active site. These typically are the carboxyl and the amino groups of polar amino acids of the Ag and Ab molecules. van der Waals forces are weak electrostatic interactions between dipolar molecules or atoms. 2 van der Waals forces and electrostatic attractions are maximal at the shortest distances. Therefore, precise juxtaposition of oppositely charged ions on epitopes and paratopes favors strong electrostatic bonding. 2 Hydrogen bonds are the result of dipole interactions between OH and C=O, NH and C=O, and NH and OH groups with binding energy of the same order of magnitude as that of van der Waals and electrostatic interactions. Their importance in Ag-Ab interactions is diminished by the necessity of a precise fit between molecules. Although there are situations with only 1 type of interaction, for most polysaccharide, glycoprotein, and polypeptide Ags, the Ag-Ab bond is a combination of van der Waals forces and electrostatic interactions. 2 Most protein antigens are multivalent, and each valency site is generally an antigenic determinant (epitope) with a completely different configuration from the other sites; a monoclonal antibody (MAb) can react with only 1 valency site of such an Ag. 391 Table 3 lists the major factors that affect antigen-antibody binding.
Main Forces Involved in Antigen-Antibody Binding. 391

The antibody-antigen bond is reversible, and its strength can be quantified. 149 Affinity is a thermodynamic expression of the binding strength of an antibody (paratope) to an antigenic determinant (epitope). 213 The affinity constant (Ka) represents the amount of antibody-antigen complex formed at equilibrium.
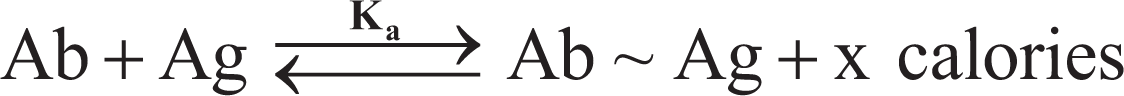
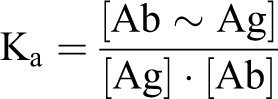
where Ab∼Ag are antigen-antibody complexes and the bracketed terms indicate molar concentrations. Ag-Ab affinity constants range from below 105 L/mol to 1012 L/mol (an Ag-Ab complex with a Ka of 1012 L/mol has 1000-fold greater affinity than one with a Ka of 109 L/mol). Affinity can be determined precisely only for monoclonal antibodies (MAbs); 213 for polyclonal antibodies (PAbs), composed of antibodies with different affinities, affinity can only be estimated. Another method currently used by antibody manufacturers to calculate the affinity of antibodies is determining the equilibrium dissociation constant (Kd). Most antibodies have Kd values in low micromolar (10–6) to nanomolar (10–7 to 10–9); very high affinity antibodies have picomolar (10–12) values. In other words, a high Ka value (eg, 1012) will correspond with a very low Kd value (eg, 10–12). New high-throughput technologies such as microarray-based kinetic constant assays 201,202 allow the Kd calculation for a large number of substances simultaneously. The affinity of an antibody for an antigen influences the sensitivity and specificity of an immunologic reaction. 149 Intrinsic affinity is determined by the sequence of amino acids of an epitope, which in turn determines the specificity of an antibody. 149 In general, high-affinity antibodies bind more antigen in less time than low-affinity antibodies, so the higher the affinity, the more dilute the antibody solution can be. 113,149
Avidity, or functional combined bond affinity, is a measure of the overall binding intensity between antibodies and a multivalent antigen. 149,213 Avidity is the result of the affinity or affinities of the antibody for the epitope(s), the number of antibody binding sites, and the geometry of the antigen-antibody complexes. 213 Thus, an antibody of IgM (decavalent) has a higher avidity (although typically lower affinity) than an antibody of IgG (bivalent) type. 149,213 High-avidity antibodies have multiple sites for secondary reagent binding, which is paramount in IHC. 213 The lower the affinity, the longer the reaction time; prolonged incubations may increase the nonspecific background. 149 The functional affinity of a PAb may be higher than that of a MAb due to its capacity to bind multiple epitopes on a single antigen. 149 To increase the functional affinity of MAbs, cocktails of antibody clones targeting the same protein are sometimes used. 149
Monoclonal and Polyclonal Antibodies
Polyclonal Antibodies
Polyclonal antibodies are produced by immunizing rabbits and various other species (mouse, goat, donkey, horse, hamster, sheep, guinea pig, rat, chicken) with purified antigen. Chickens transfer abundant IgY (IgG) into egg yolk, which makes harvesting antibodies from eggs more convenient than the typical bleeding procedure used in mammalian species. 46,213 Polyclonal antibodies have higher affinity and broader reactivity but lower specificity in comparison to monoclonal antibodies. 141 Polyclonal antisera include several different antibodies to the target protein plus, if not affinity purified, irrelevant antibodies in concentration up to 10 mg/ml. 89,237 Polyclonal antibodies are more likely than MAbs to identify multiple isoforms (epitopes) of the target protein. However, a polyclonal preparation that has not been affinity purified may be heterogeneous due to the combined presence of high-affinity antibodies and weakly immunogenic epitopes. 237 Variations in antibody titer and quality, depending on the animal immunized, also fluctuate from lot to lot. 255 The “immunologic promiscuity” of PAbs, which can amplify antigen detection by recognition of multiple epitopes, can be a disadvantage: the greater the number of different antibodies to the target protein, the greater the likelihood of cross-reactivity with similar epitopes in other proteins and, therefore, the likelihood of false positives. 237 Many proteins share immunogenic epitopes. For instance, common immunogenic domains of fibronectin III are present in at least 65 unrelated proteins. 41
Monoclonal Antibodies
Monoclonal antibodies are produced by the technique of Köhler and Milstein, 196 in which an animal, usually a mouse, is immunized with purified antigen. The specific antibody-producing B lymphocytes are then harvested from the spleen and fused with mouse myeloma cells to produce immortal hybrid cells that produce Igs specific for a single epitope (MAbs). The immortalized hybrid cells form hybridomas that are selected for the desired specificity and can be maintained in cell cultures (highly pure antibody but at low concentration) or in the peritoneum of mice (ascitic fluid with 10- to 100-fold higher Ab concentration than in cell culture, but with nonspecific proteins and extraneous endogenous Igs). One advantage of monoclonal over polyclonal antibodies is their higher specificity (Table 4). In addition, background reactivity due to nonspecific Igs is reduced (ascites fluid) or nonexistent (cell culture supernatant). The high specificity does not eliminate the possibility of cross-reactivity with other antigens because MAbs target epitopes consisting of only a few amino acids, which can be part of multiple proteins and peptides. 254 For instance, an anti–human proinsulin antibody cross-reacts with both insulin and glucagon-secreting cells. 23 Because this binding is to an identical epitope, the IHC reaction is virtually indistinguishable from that with the intended epitope. 365 It can be difficult to determine whether immunoreactivity reflects shared epitopes (cross-reactivity) or epitopes resulting from protein cross-linking during fixation with aldehydes. 141
Comparison of Polyclonal and Monoclonal Antibodies for Immunohistochemistry.

Standard Immunohistochemical Protocol Using a 2-Step Polymer-Based Detection System.a

HIER, heat-induced antigen retrieval.
aThe best incubation times for the primary antibody and other reagents should be based on information provided by manufacturers of various reagents and by personal experience.
bIncubation of the primary antibody can be done at room temperature, 37°C, or 4°C. The best incubation temperature should be determined by trial and error. The incubation chamber should have enough moisture to avoid dessication of the slides.
cSelect carefully the type of counterstain based on the chromogen used and location of the antigen.
dUse water-based coverslipping media when the precipitate of the enzymatic reaction is sensitive to organic solvents.
Rabbit Monoclonal Antibodies
The production of rabbit MAbs has been facilitated by the development of antibody libraries. 278,283,307 The advantages of rabbit over mouse MAbs include higher affinity (probably a result of high glycosylation), 149 suitability for use on mouse tissues without special procedures, increased specificity in some cases, and less need for antigen retrieval methods. 57,132,211,313,417 Nevertheless, in a study comparing 10 rabbit MAbs with 10 mouse MAbs targeting the same protein in animal tissues, rabbit MAbs were superior for some markers, but there was no significant overall improvement in immunoreactivity. 396
Protein A and Protein G
These cell-wall components of Staphylococcus aureus (A) and group G (G) streptococcal strains 46 are used in IHC for their high affinity for the Fc portion of IgGs. The IgG of many species can be bound to protein A or protein G, creating a versatile label when conjugated to enzymes or fluorochromes. Protein A/G is a genetically engineered product of Bacillus sp that combines the IgG-binding domains of protein A and protein G. Reportedly, it has higher affinity in various species than protein A or protein G alone. 46
Antibodies to Phosphorylated Proteins
These antibodies are specific for a protein activation state, so may indicate a biological state rather than the mere presence of a protein. 149,222 However, the production of these antibodies is challenging because the phosphorylation sites are at consensus sequences shared by many proteins. 149
Mouse-on-Mouse IHC
The use of mouse MAb for IHC of mouse or rat tissues results in excessive background labeling due to the binding of the secondary antibody (anti–mouse immunoglobulins) to endogenous immunoglobulins in the interstitium, plasma, B lymphocytes, and plasma cells. 49,116 This excessive background labeling has hampered or even precluded IHC with mouse MAbs in murine tissues; however, commercially available mouse-specific detection systems have eliminated this problem. One such system uses blocking steps before and after adding the primary antibody; another method preincubates the primary antibody with biotinylated anti–mouse Fab complexes (used as secondary antibody), thereby blocking free binding sites in the complexed secondary antibodies with normal mouse serum before adding the antibody mix to the tissue section. 116,152,227 Secondary antibodies to mouse IgGs may also react with Igs of other species producing the same type of nonspecific background in plasma cells, serum, or any tissue containing IgGs. The use of isotype-specific secondary antibodies, because of the low serum concentration of each isotype (20%–25%), is a relatively inexpensive method to minimize this type of background. 49
What Makes an Antibody Good for Immunohistochemistry?
Specificity and sensitivity are the most important traits. Specificity is inherent in the primary antibody. Molecular specificity, the selectivity of an antibody for a particular epitope of the target antigen, depends on its molecular structure. 149 The diagnostic specificity of an antibody against infectious agents is quantified as the proportion of samples from known uninfected animals that test negative in a given test. 419 The diagnostic specificity of an antibody against cellular/tumor markers is the expected presence or absence of immunoreactivity in certain cell types, tissues, or tumors (see Controls in Immunohistochemistry section). 369 Analytical sensitivity is determined by the least amount of antigen needed to produce a positive reaction. Polyclonal antibodies may be more sensitive than MAbs because they bind different epitopes on a single antigen. 369 Diagnostic sensitivity, the proportion of known positive samples that test positive in a given test, 374,419 is best established by comparing test results on FFPE tissue with results using another antibody validated for the same analyte or a non-IHC method, such as culture or polymerase chain reaction (PCR). Nonspecific binding of antibodies in IHC can be reduced or eliminated with reagents (including the primary antibody) of good quality; therefore, the nonspecific binding blocking step included in most IHC protocols may be unnecessary in some instances. 50
Finally, antibody selection depends on its performance in tissue sections (see standardization and validation below). As Kalyuzhny 181 wrote, “Having a good antibody for immunohistochemistry is not only about getting a strong staining signal with low background, but also about knowing the staining makes sense in terms of its histological and physiological relevance.”
Presentation of Commercial Antibodies
For a well-characterized antibody, the manufacturer’s package insert should provide all pertinent information. 368 For companies specialized in antisera production, this should include the nature of the antibody (eg, purified, whole serum, supernatant, ascites, immunoglobulin isotype), host (eg, mouse, rabbit) in which it was produced, protein concentration, immunogen used (including epitope and molecular weight, if known), species reactivity/expected reactivity (eg, human, mouse; others not known), cellular localization (eg, cytoplasmic, membrane, nuclear), recommended positive tissue controls, applications (eg, immunoprecipitation, Western blotting, enzyme-linked immunosorbent assay, immunohistology–formalin/paraffin and frozen), antigen retrieval, suggested antibody dilution, and pertinent references. Manufacturers may add client reports on species cross-reactivities. Many catalogs also include images of the immunoreaction and Western blot gels with the molecular weight of the targeted epitope(s) and the percent protein homology across species. The protein concentration of an antibody can be used to estimate the working titer; however, this is only accurate for MAbs in which the specific antibodies constitute the bulk of the reagent. For PAbs, protein concentration does not predict performance due to the presence of nonimmune proteins or antibodies with variable affinity, in addition to specific Igs. The manufacturer should be contacted for additional information on reactivity under certain conditions (formalin fixation) or species cross-reactivity if not indicated in the catalog. Data provided by only very few manufacturers in selected antibodies are their affinity constant/dissociation constant and the possibility of cross-reactivities with other antigens (eg, serotypes of viruses, other related viruses or bacteria, cell antigens) that may result from fixation or antigen retrieval procedures.
How to Find the Antibody That Matches Your Needs
Antibodies are selected based on consultation with other scientists, publications, and information from manufacturers. 47 Useful Internet resources include the Human Protein Atlas (http://www.proteinatlas.org/), which contains information on protein and subcellular profiles of numerous human cancers. 25,276,378 The Atlas lists more than 17 000 antibodies targeting proteins from more than 14 000 genes with high-resolution images to depict reactivity of the antibodies in tissue sections. Other sites with information on commercial antibodies are http://www.antibodypedia.com, http://www.biocompare.com, http://antibodyresource.com, http://www.antibodydirectory.com, and http://www.linscottsdirectory.com. Some provide information about IHC cross-reactivity and techniques for FFPE tissues or frozen sections for animal species. The South Dakota State University database is dedicated to IHC in animal samples (http://www.sdstate.edu/vs/adrdl/database/). The PHL Murine Immunohistochemistry Database (http://ncifrederick.cancer.gov/rtp/lasp/phl/immuno/) lists many biomarkers with specific protocols for IHC in mouse tissues.
Detection Systems: The Sensitivity Issue
The primary, secondary, or tertiary antibodies of a detection system are labeled with reporter molecules (labels) to allow visualization of the antigen-antibody reaction. Suitable labels include fluorescent compounds, enzymes, and metals. 218,370 The most common labels are enzymes (eg, peroxidase, alkaline phosphatase, glucose oxidase). 303 Enzymes in the presence of their substrate and a chromogen produce a colored precipitate at the site of the antigen-antibody reaction. 284
The detection system is important to maximize sensitivity of an IHC test and to optimize visibility of the immune reaction with the fewest steps and in the shortest time. 149 In addition, the detection system must be reproducible, be accurate, and render a high signal-to-noise ratio. Other factors to consider in selection of a detection system include (1) the expertise/experience of the technician, (2) type of antigens to be detected (less abundant antigens need highly sensitive methods), (3) number of tests (antibodies) available (different antibodies may require different detection systems), (4) species/tissue idiosyncrasies (eg, amount of endogenous biotin), and (5) budget. 293 Last but not least, the detection system must be compatible with the animal species. A detection system with excellent performance in human tissues may not perform well in animal tissues. There is a trend in companies marketing IHC detection systems for veterinary use to optimize their detection kits based on the species evaluated (eg, PromARK for dogs and cats, food animals, etc), reducing the chances of background or false-positive labeling. In this article, detection systems are classified as direct or indirect methods.
Direct Methods
Direct detection methods are a 1-step process using a primary antibody conjugated (labeled) with reporter molecules, 66 such as fluorochromes, enzymes, colloidal gold, or biotin. 275 The direct method is quick but lacks sufficient sensitivity for the detection of most antigens in routinely processed tissues (Fig. 11).
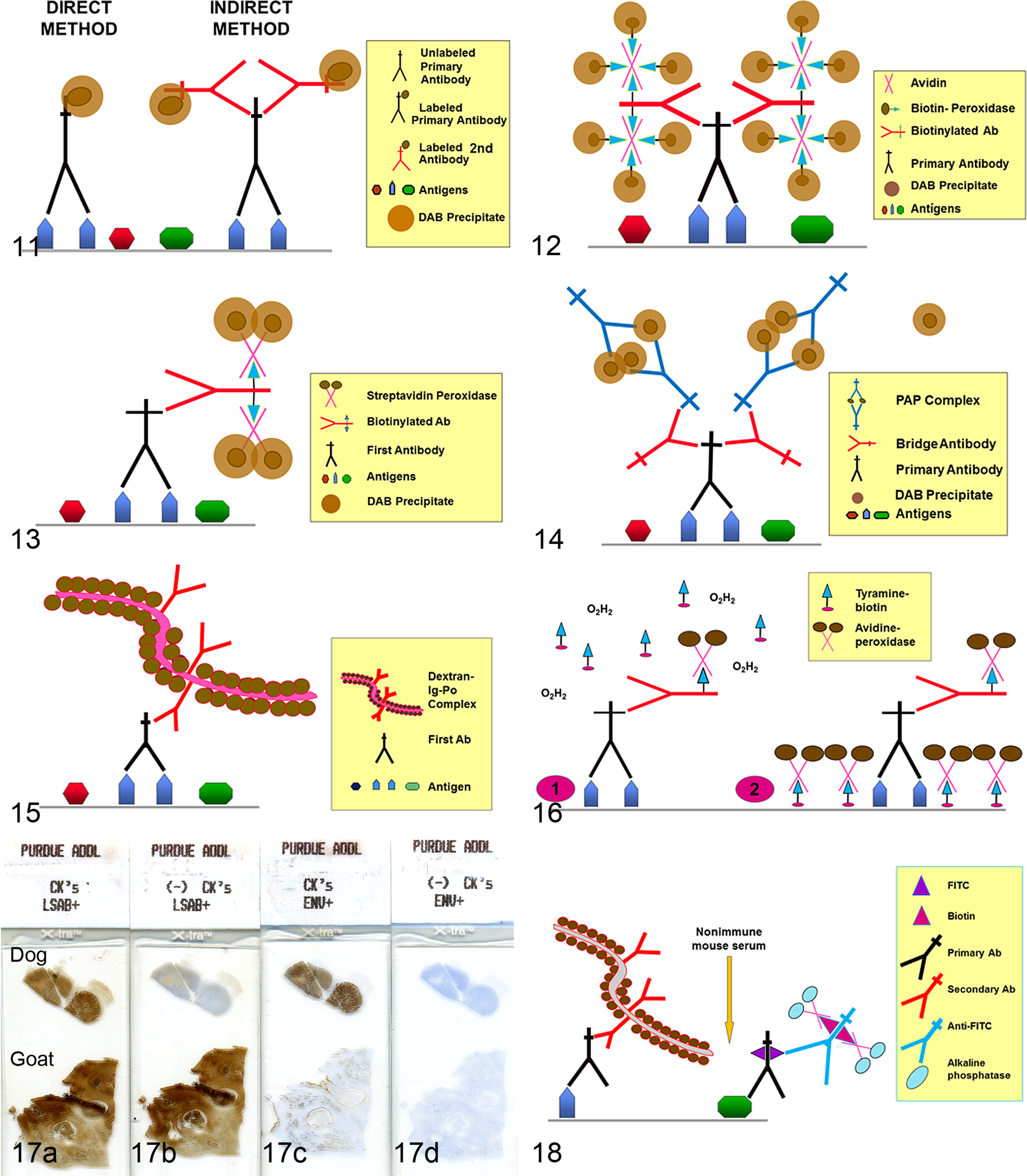
Direct and indirect immunohistochemistry (IHC) methods.
Indirect Methods
The need for more sensitive antigen detection prompted Coons et al 67 to develop a 2-step method. The first layer of antibodies is unlabeled, but the second layer, raised against the primary antibody, is labeled (Fig. 11). 275 The sensitivity is higher than with a direct method because (1) the unlabeled primary antibody retains full avidity with stronger Ag binding, and (2) the number of labels (eg, peroxidase) per molecule of primary antibody is higher, increasing the reaction intensity. Indirect methods can detect smaller amounts of antigen with less primary antibody because at least 2 labeled immunoglobulins can bind each primary antibody molecule. Indirect methods are also more convenient than the direct method because the same secondary antibody can be used to detect different primary antibodies, provided that the latter are raised in the same species. 275
Avidin-Biotin Methods
Avidin is a large glycoprotein, extracted from egg white, with 4 binding sites per molecule and high affinity for biotin. Biotin has 1 binding site for avidin and can be attached through other sites to an antibody (biotinylated antibody) or another macromolecule, such as an enzyme, fluorochrome, or other label. 275 The increased sensitivity of avidin-biotin methods reflects the larger number of biotin molecules (reporter molecules) that can be attached to a primary antibody. 134,159,160
One of the most common avidin-biotin methods is the avidin-biotin complex (ABC) method, in which the secondary antibody is biotinylated, and the third reagent is a complex of avidin mixed with biotin that is linked with an appropriate label (eg, enzyme) (Fig. 12). The avidin and labeled biotin are incubated for about 30 minutes before application, resulting in the formation of a large complex with numerous molecules of label. The proportion of avidin to labeled biotin in the third reagent must leave some sites free to bind biotin on the secondary antibody. 275 Another commonly used avidin-biotin method is the labeled avidin-biotin (LAB) or labeled streptavidin-biotin (LSAB) (Fig. 13). This method uses a biotinylated secondary antibody and a third reagent of peroxidase (or alkaline phosphatase)–labeled avidin. Its sensitivity is higher than that of the standard ABC method. 90
A disadvantage of any avidin-biotin system is the possibility of high background. Avidin can produce background by binding lectins and negatively charged tissue components through its carbohydrate groups or through electrostatic binding because its isoelectric point is 10. This background can be diminished by substituting streptavidin for avidin. Streptavidin, produced by the bacterium Streptomyces avidinii, has a neutral isoelectric point, resulting in marked reduction of electrostatic interactions with tissue. In addition, because streptavidin does not bind lectins, background labeling is less likely but still possible due to endogenous biotin. Endogenous biotin activity or tissue affinity for avidin, known as endogenous avidin-biotin activity (EABA), is particularly common in tissues that are rich in biotin, such as the liver, brown fat, adrenal cortex and kidney. Mast cells also have EABA. 138 EABA is accentuated by HIER but also develops in tissues subjected to other types of AR. 56,187,239,257,275 EABA is typically in the cytoplasm but has been reported in the nucleus. 250,257,342,428 Commercially available EABA blocking reagents (pure avidin and biotin solutions) are expensive, so many laboratories use “homemade” solutions with egg white as a source of avidin and 5% powdered milk as a source of biotin. 12,84,175,239,285,418
Non–Avidin-Biotin Methods
Peroxidase-Antiperoxidase (PAP) Method
This indirect method consists of 3 layers. 357 The third layer, which for a rabbit primary antibody is a rabbit antiperoxidase, is coupled with peroxidase in proportions to form a stable complex (peroxidase-antiperoxidase) of 2 rabbit IgG molecules with 3 peroxidase molecules, one of which they share (Fig. 14). 275 The first and third layers are bound by a bridge layer of immunoglobulins (in this example, anti–rabbit Ig). The key is to add the secondary (bridge) antibody in excess so as to bind the primary antibody through one binding site and the PAP complex through the other. The peroxidase-antiperoxidase (PAP) method is more laborious, but has 100- to 1000-fold higher sensitivity than the 2-step indirect method. PAP reagents are available for use with goat, mouse, rabbit, rat, and human primary antibodies. 370 Although PAP IHC was popular before the advent of avidin-biotin methods, its lower sensitivity limits its use today.
Polymeric Labeling 2-Step Method
This method uses a compact polymer to which 4 to 70 molecules of enzyme (peroxidase or alkaline phosphatase) plus 1 to 10 molecules of the secondary antibody are attached (Table 5). 149,411 Its advantages are (1) simplicity compared with the 3-step methods, (2) equal or higher sensitivity than ABC or LSAB methods, and (3) lack of background due to endogenous biotin or avidin. 269,318,335,370,398 However, this method is usually more expensive than ABC or LSAB methods (Fig. 15). Numerous companies have commercialized polymer-based detection systems (eg, EnVision, PowerVision, ImmPRESS, MACH 4M), which are the method of choice in many laboratories. The sensitivity can be increased with a 3-step method, in which a bridge antibody links the primary antibody and the polymer complex. Newer detection kits have replaced dextran or other macromolecules with smaller polymers (micropolymers) to increase access to the target antigen, resulting in higher sensitivity, lower background, and reduced nonspecific binding. 149
Catalyzed Signal Amplification
This method is based on the ability of tyramide to adhere to a solid substrate (eg, tissue section) following oxidation/radicalization. 131 The procedure, adapted for IHC, 3 is based on the deposition, catalyzed by horseradish peroxidase (HRP), of biotinylated or FITC-conjugated tyramide at the location of the antigen-antibody reaction. Free radicals formed during the HRP-tyramide reaction bind covalently to electron-rich amino acids (eg, tyrosine) of nearby proteins. 48 The reactive intermediates are so short-lived that biotinylated tyramide is deposited only at or near its site of production. 143 The biotin conjugated to the tyramide subsequently complexes with avidin conjugated to HRP. 143 This method is complex and laborious because it involves an initial avidin-biotin procedure followed by the tyramide reaction (Fig. 16). However, its sensitivity is 5- to 10-fold higher than that of the regular avidin-biotin method; 350 others claim an even greater increase in sensitivity. 234 This method is suitable for antigens present in very low amounts. However, background can be a problem, particularly when using HIER, 140 so prolonged treatment to quench endogenous peroxidase or EABA is usually necessary. Modifications using fluoresceinated tyramide result in marked reduction or ablation of background from endogenous biotin. 139,140,259,389
Immuno-Rolling Circle Amplification
Rolling circle amplification (RCA) increases the signal of the immunologic reaction without increasing the noise (background) that can occur with the tyramide amplification method. Rolling circle amplification is a 2-part, surface-anchored DNA replication used to visualize antigens (immunoRCA). The first part is an immunologic reaction (antigen-antibody binding); the second part is an isothermal nucleic acid amplification using a circularized oligonucleotide primer. 216,325,436 The primer is coupled to the antibody, so in the presence of circular DNA, DNA polymerase, and nucleotides, the rolling circle reaction results in a DNA molecule consisting of multiple copies of the circular DNA sequence that remain attached to the antibody. The amplified DNA can be detected by hybridization with labeled complementary oligonucleotide probes. 325 The main differences between RCA and PCR is that the former can amplify nucleic acid segments in either linear or geometric kinetics under isothermal conditions, and the product of amplification remains connected to the target molecule. 216,325 The linear mode of RCA can generate 105-fold signal amplification during a brief enzymatic reaction. ImmunoRCA is sensitive enough to detect a single antigen-antibody complex. 325
Polyvalent Detection Systems
Many manufacturers offer polyvalent (sometimes called universal) detection systems. They differ from 1-species detection systems in that the secondary reagent is a cocktail of antibodies against immunoglobulins from different species, allowing 1 secondary reagent to be used for both polyclonal (eg, rabbit and goat) and monoclonal (eg, mouse) antibodies. These systems reduce the complexity of IHC; however, due to the “universal” recognition by the polyvalent antibodies, awareness of potential species incompatibilities is critical (Fig. 17).
Increasing the Sensitivity of Antigen Detection
The more sophisticated and highly sensitive detection methods can be prohibitively expensive. Therefore, standard methods (eg, PAP, ABC, LSAB) have been modified by combining methods, repeating steps of the immune reaction, increasing the incubation time of the primary antibody, or enhancing the intensity of the chromogen precipitate. §
Detection of Multiple Antigens in a Tissue Section
Multiple immunolabeling is used to localize different antigens (eg, cell markers plus infectious organisms) in the same section. The availability of various detection systems (PAP, ABC, polymer-based methods, commercial dual-labeling kits), enzymes, and chromogens with different-colored reactions makes multiple immunolabeling feasible. A potential pitfall is false-positive labeling due to cross-reactivity among different components of the reaction. This requires careful planning and the use of multiple controls. Double immunolabeling methods can be simultaneous (parallel) or sequential. 382 Simultaneous double immunolabeling is performed with primary antibodies made in different species; both primary antibodies are added simultaneously, followed by a mixture of different enzyme-conjugated secondary antibodies and subsequently developed with the appropriate chromogen/substrate solutions. 149 As a rule, if the primary antibodies are from the same species, sequential dual labeling is necessary. The risk with sequential dual labeling is that the second layer of antibodies (intended for the second antigen) can also bind the first antigen through the primary antibody (Fig. 18). Therefore, an intermediate step is inserted between the first and second IHC reactions to elute the primary antibodies of the first reaction with potassium permanganate or a solution of glycine-HCl for several hours or HIER. 89,384,387 The use of glycine-HCl with SDS at pH 2 as an elution agent for sequential dual labeling did not reduce immunoreactivity. 274 The elution step could be omitted by developing the first reaction with a concentrated diaminobenzidine solution, which theoretically would block any residual primary antibody of the first immunoreaction. 89 There are commercial kits with an elution or HIER step between the first and second immunologic reactions. 382 Sequential double-labeling techniques are not recommended if mixed-colored products as a result of colocalization of antigens are expected. 382 In other words, sequential dual labeling is more appropriate for the detection of antigens in 2 different cell populations or cell compartments (eg, nucleus and cytoplasmic membrane). A novel IHC procedure can simultaneously visualize 5 biomarkers within a single tissue section using the same chromogen (amino-9-ethylcarbazole [AEC]) and antibody elution with KMnO4 and H2SO4. 122
Multiple immunolabeling requires stringent controls and careful combination of enzymes and chromogens to achieve the best color discrimination (contrast) of the IHC reaction. The optimal order of antigen detection depends on several factors, including the need for AR. The choice of counterstain depends mainly on the color of the immune reaction; it should lightly stain the tissues and differ sufficiently in color from that of the chromogen precipitate to avoid confusion. The most frequently used counterstains are hematoxylin, methyl green, and nuclear fast red. 390 In some situations, particularly with nuclear antigens in small amounts, counterstaining is not recommended. 284
The Color of the Antigen-Antibody Reaction
The antigen-antibody reaction is not visible under the microscope unless labeled. The most common labels are enzymes, particularly peroxidase and alkaline phosphatase. Each enzyme has specific substrates and chromogens to produce a colored precipitate. The common chromogens impart a brown, red, or blue color to the reaction. The choice of enzyme and chromogen depends on several factors, such as the reaction intensity, antigen location, presence or absence of endogenous pigments, or mounting media used, but often is a matter of personal preference. 390 Many laboratories prefer peroxidase, but alkaline phosphatase (AP) is also commonly used and is claimed to be more sensitive than similar immunoperoxidase methods. 229,230 For horseradish peroxidase, 3,3′-diaminobenzidine tetrachloride (DAB) is the usual chromogen, imparting a brown color that is insoluble in organic solvents. 284 When endogenous peroxidase activity is high or melanin pigment is prominent, other chromogens or alkaline phosphatase should be used. 370 3-AEC, another chromogen for peroxidase, produces a red color. However, coverslips must be mounted with a water-soluble medium because AEC precipitate will wash out with organic solvents. 4-Chloro-1-naphthol forms a blue precipitate that is also soluble in organic solvents. For alkaline phosphatase IHC, 5-bromo-4-chloro-3-indolylphosphate/nitro blue tetrazolium chloride (BCIP/NBT) (blue, permanent media), fast red (red, aqueous mounting media), and new fuchsin (fuchsia, permanent media) are the usual chromogens. 284 Alkaline phosphatase is recommended for immunocytochemistry. 31 Van der Loos 385 reviewed the chromogens used in multiplex IHC with photonic microscope and spectral analysis. Newer chromogens are more stable and produce a wider range of colors of the final product.
Multispectral Analysis
Selecting the right set of substrates for multiplex IHC can be difficult, particularly when antigen colocalization causes mixing of the colored reaction. A novel alternative approach to photonic multiplex IHC is multispectral analysis, in which the colors produced during IHC are converted to a spectrum analyzed by computer. 105,384 Multispectral analysis is also semiquantitative. 384
Quantum dots (QDs) are semiconductor nanoparticles that can be used in multiplex IHC. QDs emit different wavelengths depending on their size (1–10 nm), shape, and composition. 174,268,270,430 QDs can be linked to antibodies, oligonucleotides, aptamers, and streptavidin for specific binding to tissue biomarkers 430 and are particularly useful to study tumor heterogeneity. 174,214,215 Advantages of QDs in comparison to standard organic fluorochromes include narrow emission spectra (less interference in multiplex analysis), higher photostability (at least 10 hours), and higher fluorescent signal per unit of light absorbed. 98,268,430 In addition, QD IHC is more accurate and precise at low protein concentration than traditional IHC. 61
Refined techniques combining multispectral analysis and tissue microarrays have made high-throughput biomarker quantification and colocalization more accurate and less cumbersome. 102,163,226 These techniques are described in more detail in another article. 219
Manual vs Automated Immunohistochemistry
Laboratories performing IHC on a routine basis benefit from automated systems. Automated IHC follows the same procedure as manual IHC, but the procedure is programmed into a computer that controls the steps executed by the autostainer. Automated IHC saves technician time and typically produces more consistent results. It has evolved from rudimentary machines to refined models that can perform the entire procedure, even multiplex IHC, without human intervention.
Selection of the best autostainer depends on the laboratory’s needs in terms of the number of IHC tests, procedural complexity, and whether the same platform is to be used for other types of testing (eg, in situ hybridization). Once the needs for IHC are defined, the choice of platform may be reduced to a few commercially available instruments. This review does not address the advantages/disadvantages of each automated IHC platform but focuses instead on the factors to consider before purchasing an autostainer.
The number of slides that can fit in an autostainer is critical. Small units accommodate 20 to 30 slides at a time. Additional units can be connected and controlled by the same computer, providing flexibility if demand for testing increases. “Closed” systems versus “open” systems is one of the most hotly debated choices in IHC platforms. With a closed IHC platform, most reagents and consumables (pipette tips, slide holders, etc) must be purchased from the platform’s manufacturer. With an open system, reagents (including glass slides) from other companies can be used without affecting the quality of the IHC reaction. In reality, some platforms classified as “open” are only a bit more open than a closed one. Closed systems are favored by laboratories that run numerous IHC tests on human tissues with little time for tweaking procedures. It is precisely because most IHC reagents have been developed for use in human tissues that the closed system has limited appeal in veterinary medicine, except for certain regulatory tests (eg, prion testing), in which numerous samples are run simultaneously using a procedure validated for a particular IHC platform. For routine veterinary IHC, an open system allows the necessary flexibility to perform IHC in a wide variety of animal species with both commercial and homemade reagents.
“Online” vs “Offline” Antigen Retrieval
Some platforms allow HIER as a part of the IHC procedure. 251 Although this feature provides greater flexibility, at least with standard AR, these platforms are more expensive and require higher maintenance that those without “online” HIER. Several platforms are designed for continuous access, meaning that, as some tests are finalized and the slides are removed from the stainer, a new batch of slides can be added without stopping the IHC run. This feature is valued in human clinical laboratories with high demand for rapid testing. Other platforms, especially those with ISH capability, can use higher incubation temperature (eg, 37°C) to increase reaction speed. Reagent volume may determine the cost of a test; some platforms require about 100 μl reagent per slide regardless of the surface area to be covered, whereas others need up to 3 times more reagent. The need for special devices to allow incubations with smaller reagent volumes should be considered in the final cost.
Perhaps the most important factor in selecting an autostainer is the company behind it—in other words, the quality of technical service and responsiveness of the company to the laboratory’s needs. The most sophisticated machine is of little value if the service provided by the manufacturer is suboptimal. It is uncommon, particularly in veterinary medicine, for the manufacturer’s software package to satisfy all the needs of a particular laboratory. The willingness of a manufacturer to customize the software to the individual laboratory’s needs is important in purchasing decisions. Calculation of the cost per slide produced with an autostainer must include the annual/semiannual maintenance service fee. As Rodney Miller, “Although automated immunostainers have improved the quality of immunostains in many laboratories, the machines are not perfect, and it is naïve to assume that obtaining an automated instrument will guarantee quality and reproducibility.” 239
Storage and Handling of Reagents
The shelf-life of IHC reagents varies depending on their nature and storage conditions. Many primary antibodies and detection kits can be used beyond the manufacturer’s expiration date with proper handling and storage. 11,323,377 However, such use must be properly documented.
Diluent Buffer
Antibodies are attracted to the epitopes of most antigens initially through electrostatic charges and subsequently through van der Waals and hydrophobic interactions. 32 The isoelectric point (pI) of polyclonal IgG ranges from 6.0 to 9.5, so the antigen-antibody reaction is relatively stable at pH 6.5 to 8.5 when using whole sera as primary antibody. Although IgG MAbs have a similar pI range, 143 slight fluctuations in pH can adversely affect antigen binding. 2,427 Reportedly, 408 MAbs perform better in phosphate-buffered saline (PBS) than in Tris buffer. This is puzzling because sodium ions (responsible for the ionic strength of a buffer) tend to shield negatively charged epitopes, thereby diminishing the attraction to positively charged paratopes of the antibody, especially with alkaline buffers. 143 Phosphate ions, on the other hand, promote hydrophobic binding, which could explain the superiority of PBS in some instances. Many authors conclude that the best diluent buffer for MAbs and PAbs is 0.05 to 0.1 M Tris buffer (pH 6.0), but there are exceptions. 31,33,429 Tris buffer should also be used for alkaline phosphatase procedures, because the high concentration of inorganic phosphate competes for the phosphatase. 89 Background reactivity due to ionic interactions can be reduced by increasing the NaCl concentration in the buffer to 0.3 to 0.5 M; 277 however, increased ionic strength can disrupt the binding of specific but low-affinity antibodies, so it should be used judiciously.
Assay Standardization
Standardization determines the optimal conditions (eg, incubation time, incubation temperature, dilutions, pretreatment techniques, controls, buffers, detection system) for an IHC protocol. In IHC, the goal of standardization is reproducible and consistent results within each laboratory and comparable results among laboratories even with different procedures. 291 Unfortunately, with the exception of the surveillance program for prion diseases, neither the protocols nor the primary antibodies are uniform among diagnostic laboratories. This lack of standardization of IHC tests among veterinary laboratories 284 is complicated by the scarcity of high-quality antibodies designed for the infectious agents and cell markers of animals. 291 Particularly in tumor diagnostics, most veterinary laboratories use batteries of antibodies developed for use in human tissue.
Even in human medicine, there is little guidance or regulation for standardization of IHC tests, although a consensus on guidelines for quality assurance for IHC has been published by the CLSI, formerly the National Committee on Clinical Laboratory Standards. 149 Based on the assumption that most IHC tests are part of the pathologist’s report and not reported separately, 291 the Food and Drug Administration (FDA) has classified most IHC reagents and kits as “Analyte Specific Reagents,” thereby exempting them from premarket notification. For antibodies used as “stand-alone tests” in human pathology, premarket notification, and specific FDA approval are required. For some stand-alone tests in veterinary IHC (e.g., detection of persistent bovine viral diarrhea [BVD] virus infections), premarket notification by regulatory agencies (US Department of Agriculture [USDA]) is not required. However, for the transmissible spongiform encephalopathies, in which IHC is a stand-alone test for the prion protein, the USDA mandates a standardized procedure, depending on the IHC platform, among laboratories.
Practical Standardization of a New Antibody
Tissue preservation affects performance of the primary antibody. Frozen sections typically require less incubation time than FFPE sections and, depending on the fixative, may not require AR. Although several commercially available fixatives reportedly preserve epitopes for IHC better than formalin, they are more expensive and often less suitable for routine microscopy. 291
Primary antibody characterization
The use of a primary antibody in a species other than the intended one requires scrutiny of the specification sheet and additional testing. A Western blot can be used to demonstrate reactivity with the appropriate molecular weight antigen in a particular species; however, Western blot immunoreactivity does not necessarily predict immunoreactivity in FFPE tissues. Comparison of immunoreactivity with that in the “gold standard” species is necessary, but antigen distribution can vary among species. For viruses, immunoreactivity should be detected in the appropriate tissues, cells, and cellular location, as well as in association with typical lesions. For protozoa and bacteria, consistent morphology and location of the labeled organisms supports IHC results. For cell markers, reactivity should be detected in the appropriate cell or tissue compartment (Fig. 19). More information on primary antibody evaluation is in the section on test validation.
Each antigen has a typical anatomic compartment. (a) Carcinoma, mouth; dog. Cytoplasmic pattern for cytokeratins. (b) Lymphoma, lymph node; dog. Cell membrane location of CD20. (c) Histiocytic sarcoma, spleen; dog. Although CD18 is a cell membrane antigen, it may be expressed in a paranuclear (Golgi) location. Inset: Detail of the reaction. (d) Plasmacytoma, skin; dog. The transcription factor MUM1 is expressed in plasma cell nuclei. Inset: Detail of reaction. Immunoperoxidase-DAB, hematoxylin counterstain.
Antibody dilution
The optimal dilution of the primary antibody is that titer with the highest ratio of “signal” (specific reaction) to “noise” (background labeling) (Fig. 20). 149 The optimal titer should produce appreciable differences in labeling intensity within and among samples such that strong- and weak-positive reactions are distinct from the negative reaction. 149 This dynamic range of labeling is particularly important in scoring immunoreactivity for prognostic or therapeutic decisions. 149
Primary antibody dilutions for one type of tissue may not be optimal for another type. The ionic strength of the diluent can be adjusted to improve the signal-to-noise ratio, particularly for PAbs in liver sections. 431 In a diagnostic setting, however, the optimal working titer should accommodate a range of relevant tissues. Tissue processing for IHC must be standardized to match that used in standardization tests to avoid effects on the optimal titer.
The manufacturer’s data on antibody performance in FFPE tissue can be used as a guideline for the starting dilution. Depending on the antibody type (monoclonal or polyclonal) and quality, the working concentration should be 1 to 5 μg/ml and can be reduced with AR procedures. 47 Secondary antibodies (working concentration, 5–10 μg/ml 47 ) and detection complexes in commercial kits do not need to be titrated. 291
To determine optimal dilution of a new antibody, the following 3 sets of 5 slides each are recommended: 1 set without AR, 1 set with enzymatic AR, and 1 set with HIER. The first 4 slides in each set have sequential 2-fold dilutions of the primary antibody (typically, the midrange dilution is that recommended by the manufacturer); the fifth slide is incubated with the negative reagent control (Fig. 21). 54,287,291,303 A similar approach is recommended by the College of American Pathologists. 157
Incubation conditions
The conditions for incubation of the primary antibody depend on its affinity, ambient temperature, section characteristics, and procedure. 291 Incubation for most routine protocols is 30 to 90 minutes at RT. Incubating sections must be completely covered by an adequate volume of the antibody solution to ensure even exposure of the tissue and prevent drying. Shorter incubation at 37°C to 40°C, longer incubation at RT, or extended (overnight) incubation at 4°C can optimize immunolabeling. 37 Slides should be incubated in a chamber to prevent evaporation of the antibody solution. 149 For longer incubations and to reduce evaporation, a container of buffer or water can be placed within the incubation chamber. 149
Many antibody traits affect incubation time. Specificity is important in selecting the best duration of incubation. Lower affinity antibodies require longer incubation times, higher concentrations, or higher incubation temperature. 291 Different lots of the same antibody may vary in concentration or other characteristics and thus require re-titration.
Interspecies variations in IHC reactions result from subtle changes in the amino acid sequence of the targeted antigen (Fig. 22). 290 Even with proven interspecies cross-reactivity, the antibody affinity may be decreased in a different species. Prolonged incubation, adjusted AR, and/or increased antibody concentration may be needed for optimal IHC. Antibody specificity must be verified in each species tested. 291
Choice of detection system
Many commercial detection kits, some optimized for particular animal species, claim to be the most sensitive, but any detection kit must be validated in house before routine use. The secondary and tertiary reagents of these kits may contain antibodies or other compounds that could react nonspecifically with tissue antigens, increasing background or producing false-positive results. This is one justification for negative controls in IHC (see below in validation). Typically, a non–biotin-labeled polymer detection system is recommended if HIER is used to avoid background from EABA. 54
Postanalytical Phase of IHC
Assay Validation
Validation is a process to affirm the suitability of an assay, once it has been developed, optimized, and standardized, for a specific purpose. 419 Validation appraises the analytical and diagnostic performance of a test. 419 A validated IHC test should be reproducible with high sensitivity and specificity. 71 Validation entails several steps starting with evaluation of analytical characteristics: specificity and sensitivity, repeatability, and preliminary reproducibility. 419 Next, diagnostic characteristics, particularly diagnostic specificity and sensitivity, and cutoff value are evaluated. Finally, the test’s reproducibility and ruggedness are evaluated with implementation among laboratories and continuous monitoring of validation criteria. 419 Test validation requires proof of antibody specificity in an established assay. For most IHC tests, this involves detecting any cross-reactivity of the antibody with unrelated antigens (or the presence of antigen in a cell or tissue not previously known to harbor it) or among different tissues species. Moreover, validation examines the variables that affect the IHC reaction, such as fixation time and storage of paraffin blocks or unused tissue sections. 291 A single assay may be validated for several purposes by optimizing for each intended purpose. 419 For example, if an IHC marker (analyte) is to be used to demonstrate the same antigen in different animal species, antibody dilution, incubation time, AR, and other test parameters may need adjustment. An IHC test can be validated by comparing results among laboratories using similar techniques. 16 When possible, validation compares the IHC test sensitivity to that of the “gold standard” method for the antigen in question. It may also compare staining patterns and sensitivity with other antibodies targeting the same antigen. Test validation follows test standardization. 419 The CLSI evaluation protocol for a qualitative diagnostic test, such as IHC, in situ hybridization and Western blotting analysis of 50 positive and 50 negative specimens if validation data are not available from other sources (eg, antibody manufacturer). 71 This target can be difficult to achieve in veterinary medicine where validation may have to be an ongoing process. The in-house validation procedure depends on the antibody and the laboratory. A validated IHC assay should produce consistent results, regardless of the method or the laboratory where it is performed. 291
As mentioned earlier, validation of an IHC test may require an additional method (eg, the gold standard) to prove the presence or absence of the antigen in question. In some cases, the gold standard method does not prove the presence of a protein but demonstrates a physiological activity closely associated with the given protein. An example is the validation by Martinet et al 228 of an assay to demonstrate autophagy-related proteins. The gold standard to detect autophagy is transmission electron microscopy (TEM). However, because this method is time-consuming and not routine, the authors developed an IHC assay. Autophagy was induced in mice by starvation (positive control, confirmed by TEM); the negative control was tissue from transgenic mice with the essential autophagy gene Atg7 deleted. These mice lacked autophagolysosomes by TEM, even when starved. Different commercial primary antibodies, putatively specific for autophagy, were used as well as different fixation procedures, buffers, and AR protocols. The use of Atg7 knockout mice as a negative control proved that some of the tested antibodies indeed recognized a different protein in normal and starved mice.
The use of acetone- or ethanol-fixed frozen tissue sections has been advocated as the gold standard against which IHC on FFPE should be compared. However, in some instances, frozen sections fixed in coagulating fixatives are suboptimal for IHC compared with FFPE tissue sections. 338 This is particularly relevant when testing low-molecular-weight proteins and lipoproteins, which are readily extracted by coagulating fixatives. 203 The same conclusion was reached by van der Loos 383 in studying the distribution of interferon (IFN) γ–producing cells using different fixatives (Fig. 6). This point is corroborated by international governing organizations of diagnostic assays, which indicate that the matrix (eg, tissue section) in validation studies should be identical or closely resemble the samples to be tested. 419
Primary Antibody Characterization
Validation of an assay requires confirmation that the primary antibody is specific, selective, and reproducible for its intended use. 40 For cellular markers, a good starting point is Western blot (WB) to demonstrate reactivity with the appropriate molecular weight antigen in the tissue and species of interest; 320,386 multiple bands or bands at inappropriate molecular weight could indicate posttranslational modifications or low antibody specificity. 40,200,321 Nevertheless, WB immunoreactivity does not necessarily predict immunoreactivity in FFPE tissues; WB requires antigen denaturation (therefore, modification of its secondary and tertiary structure) as opposed to the in situ antigen modification in formalin-fixed tissues, with cross-linking rather than denaturation. 55,413 Some antibodies bind only denatured protein immunoblots; others bind only native proteins. 55,69 In some cases, as with HIER, the antigen denaturation by WB may not be problematic and does not disqualify this procedure, because IHC detection of most epitopes requires only an intact primary (linear) protein structure. 39,347
Incubating the antibody with excessive blocking peptides before the IHC reaction (the so-called preadsorption test) has been used in validation. 40,155 However, blocking peptide procedures do not prove antibody specificity or provide information regarding signal-to-noise ratios, 360 and the amount of antigen that constitutes an “excess” can only be calculated by knowing the Kd of the antibody. 321 Diluting the antibody to the point that labeling occurs only in expected locations is a more logical approach. 321
Despite their shortcomings, WB and preadsorption tests are the most widely used methods to determine the specificity of a primary antibody in a diagnostic setting. Other methods that require more sophisticated technology and laboratory capabilities include (1) use of knockout animal tissues to demonstrate the lack of detection of the protein genetically removed from that animal and (2) use of a transfected cell line expressing the antigen for the primary antibody. 55,320 Comparison of immunoreactivity with the “gold standard” species is another option; however, antigen distribution may vary among species. 291 In the end, antibody specificity is best documented by the appropriate use of controls (see below). 40
A special situation arises with commercial purified affinity MAbs that claim specific antigen targeting as demonstrated by WB on a limited set of tissues or cell culture extracts. By definition, these antibodies are specific for a particular epitope. In reality, some of these “commercially validated” antibodies cross-react with unrelated antigens associated with degeneracy and immune mimicry. 63,279 This unexpected cross-reactivity is difficult to detect without extensive antibody validation with different tissues by WB or paraffin sections. 386 For most laboratories, this approach is neither practical nor economically feasible.
A common practice in the biopharmaceutical industry is tissue cross-reactivity (TCR) studies to identify off-target binding of primary antibodies or antibody-like molecules with a complementary-determining region; TCR is also used to detect previously unidentified sites of on-target binding. 204 These pharmaceutical compounds, intended as vehicles of biologically active molecules, are incubated with microarrays of human or animal tissues (frozen or, less commonly, fixed); TCR studies supplement but do not replace in vivo studies. 204
In some laboratories, the same MAb (same clone and antibody presentation) from 2 different companies is used in the same IHC procedure for validation purposes. We have experienced aberrant immunoreactivity using an antibody to CK8/18 from one company, whereas the same clone from a different company produced the expected results (Figs. 23, 24). This discrepancy suggests that nonactive ingredients of the monoclonal preparation could have interfered with the immunologic reaction.
Manufacturer differences in MAbs. Intestine; dog. MAb 5D3 cell culture supernatant for cytokeratins (CK) 8/18. Compare with Fig. 24, from a different manufacturer. (a) Expected reactivity in mucosal epithelial cells (solid circle) with no labeling of mesenchymal cells in the submucosa (s) or muscularis (m). (b) Detail of the mucosa. (c) Detail of the submucosa and muscularis. Immunoperoxidase-DAB, hematoxylin counterstain.
Cross-Reactivity
Cross-reaction can result in a false-positive reaction. 419 However, in IHC, 2 main types of cross-reaction, neither of which necessarily implies a false-positive reaction, can be used to the diagnostician’s advantage within the proper context: antigen cross-reactivity and species cross-reactivity. It is assumed that test conditions are those used during standardization. 291
Antigen cross-reactivity
The structure and amino acid sequence of an antigen affects its cross-reactivity. A literature search may reveal reports of antigen cross-reactivity. Lack of cross-reactivity in one species does not preclude it in another. 291 Molecular mimicry (eg, cross-reactions of infectious agent antigens with normal tissue antigens) may also occur. 353
Evaluation of cross-reactivity in the diagnostic setting depends on the category of the antibody (against infectious agents vs cell markers). 291 Antibodies against bacteria, protozoa, or fungi should be tested against other microorganisms that are morphologically similar, belong to the same group, or produce similar lesions in the organ of interest. Antibodies to viruses should be tested against other viruses in the same group that affect the same tissue or produce similar lesions. Some lack in specificity does not preclude the use of an antibody, provided it is documented and interpreted. For example, an antiserum against porcine coronavirus is used to detect feline enteric coronavirus, feline infectious peritonitis virus, or mink and ferret coronaviruses. Some MAbs against infectious agents cross-react with cellular organelles.
For neoplasms, antibodies to specific cell types or components should be evaluated in 1 or more of the following ways: (1) comparison of immunoreactivity in primary versus secondary (metastatic) tumors (Fig. 25), 288,297 (2) determination of immunoreactivity in other neoplasms in the differential diagnosis, 289,294,296,297,299,302 (3) comparison of immunoreactivity between nonneoplastic and neoplastic tissues (antigen expression may be lost or expressed de novo in neoplastic cells) 301 or between benign and malignant phenotypes, 392 (4) evaluation of immunoreactivity in tumors with different histologic patterns or cellular phenotypes, 288,295,297 (5) determination of antibody cross-reactivity between neoplastic and nonneoplastic cells in the organ of interest (eg, hepatocellular tumors vs secondary hepatic tumors), 298 (6) comparison of reactivity between wild-type and mutated antigens (eg, p53), and (7) semiquantitative evaluation of immunoreactivity of various antibodies targeting the same cell or cell product in various tumors or tissues to determine their relative diagnostic utility (Fig. 26). 290,301,302
Species cross-reactivity
Although some antigens can be detected under similar conditions in different species, it should be assumed that antigen detection varies among species. 301 Identical antibody clones differ in reactivity among species. Not all antibodies targeting particular antigens will be reactive in all species. For example, Melan A labels melanocytes in dogs, cats, and other species but not in horses. 290 It is advisable to restandardize (optimize) an IHC test for each new species, including incubation times, concentration of the primary antibody, and AR. A literature search or consultation with the antibody manufacturer and extensive testing of similar cases in a given species are recommended to determine species cross-reactivity. 291 A diagnostic IHC report should indicate the degree of confidence in the results based on experience and publications. The use of tissue arrays from different animal species permits rapid screening for interspecies cross-reactivity. 173,265,284
Effects of Fixation, Postfixation Treatments, and Equipment on Immunoreactivity
These laboratory variables affect assay development and validation. 419 Different fixatives have different effects on immunoreactivity. For each fixative, full standardization of a test must be done, including incubation conditions, dilution of the primary antibody, and AR method (see section on standardization). Standardization is usually done on tissues with known fixation time (eg, 1–2 days).
Fixation kinetics studies are recommended for each antigen or epitope as part of the validation process to determine the assay robustness, defined as its capacity to produce reproducible results despite small variations in method parameters (eg, incubation temperature, pH of buffers, batch of reagents, shelf lives and storage conditions, fixation length). 419 A positive tissue control should be fixed for different durations (eg, 1, 2, 4, 7, 10, 14, 21, or 20 days) and tested under identical conditions to determine the effects of fixation time on reactivity (ie, intensity and number of cells or organisms detected (Fig. 3). 288,297,403,404
Automated stainers are designed to duplicate manual procedures and ensure uniform application of each step of the process. Thus, the use of automated equipment creates a uniform and standardized testing environment, which will improve intralaboratory run-to-run consistency. 251,369,433 When results among different laboratories diverge considerably, technical differences should be considered as a possible cause.
What Constitutes a Positive/Significant Result?
There is no single answer to this question. IHC assays detect analytes (infectious agents or tumor marker antigens) in tissues, but diagnosis requires interpretation of IHC results in the context of clinical, pathologic, or laboratory findings (the so-called class I IHC tests). 375 For an infectious agent, detection of even 1 organism indicates infection, but it is another matter to prove that the agent caused disease. For neoplasms, interpretation is even more complex, because tumors are often heterogeneous, and tumor stem cells can differentiate in various directions. The reported percentage of positive cells required to confirm tissue origin of a tumor varies considerably. A cutoff value of 10% positive cells has been suggested to classify an IHC test as positive, 375 but the cutoff value is by no means set in stone. 136 Some pathologists prefer the words detected or not detected rather than positive or negative, which underscores the need for subjective interpretation of the IHC reaction in the context of the disease. 326 Each antigen has a “different weight” in the final interpretation. A more pragmatic approach would be to set a cutoff value for each antibody, optimized for a given entity. For example, the cutoff value for CD34, consistently expressed in many different mesenchymal cells, should be higher than that for uroplakin III, which, although highly specific for urothelial tumors, may be expressed in only a small proportion of the neoplastic cells. 297 For prognostic markers or those used to customize therapy, a standardized scoring system applicable to different platforms is required for meaningful results. 380,402
How to Distinguish True-Positive From False-Positive Immunoreactivity
The pattern of immunoreactivity, particularly with neoplasms, is important in distinguishing a true-positive reaction, which has some degree of cell-to-cell heterogeneity, from a false-positive reaction, which lacks heterogeneity, even in the absence of stromal background reactivity. 239 Reactivity of a biomarker in tissues/cells not reported previously is not unusual; less commonly, an antigen is detected in a location not consistent with its function. For example, thyroid transcription factor 1 (TTF1) expression is expected in the nucleus, but not cytoplasm, of thyroid and pulmonary neoplasms; 299,300 however, cytoplasmic TTF1 expression has been reported in human liver neoplasms and confirmed in mitochondria by other methods. 58,267,410 Whether the detected antigen is part of the TTF1 molecule or a cross-reacting antigen is not clear.
Clinical Validation
Clinical validation is based on whether a test result correlates with case outcome or response to therapy. 126 Clinical validation is common in human oncology but requires access to hospital medical records from numerous cases. In veterinary medicine, prognostic or therapeutic assumptions are often based on published information. 24,191,319,373,395
Controls in Immunohistochemistry
Positive Tissue Control
A positive tissue control is a tissue known to contain the targeted antigen detectable by identical IHC methods to those used in diagnostic cases. 149 Positive tissue controls assess the performance of the primary antibody. 149 Fresh-fixed surgical biopsy specimens are preferred over postmortem specimens, because extended warm ischemia and autolysis affect immunoreactivity. 149 For laboratory animals, autolysis is less of an issue, and postmortem specimens are routinely used. Positive tissue controls must be fixed, processed, and stained in the same way as the test tissue for each antibody and procedure. 306,369 Control tissues should be from the same species as the test tissue, with few exceptions (see below). The antigen in the positive control should not be abundant and should have some heterogeneity in intensity throughout the sample; weakly immunoreactive areas can be used to detect subtle changes in antibody sensitivity. 369 A positive control with abundant antigen may reduce the chances of identifying weak-positive cases. 149 The presence of the antigen in the tissue control should be confirmed by another method (eg, PCR, virus isolation) or documented by publication. For most laboratories, control tissues are generally obtained “in house” because fixation and processing procedures are those used with the test tissue. Control tissues from another laboratory or a commercial source could have been fixed or processed differently. When possible, the positive tissue control should be on the same slide as the test tissue to minimize technical variation.
Multitissue blocks and cell lines
Various methods for the simultaneous study of multiple FFPE tissues in a single histologic section have been reported. In the sausage technique, 17 long thin strips of fixed tissues are drawn into a tube of unfixed small intestine. The entire sample is then fixed, resulting in a tight column that can be cut into blocks and embedded. Substitution of placenta as the “wrapping” tissue 115 and other modifications 239 to the original method have been reported.
Paraffin-embedded tissue microarrays
These have been used as controls in human IHC laboratories, mainly for tumor diagnosis. 158,265,266,284 Validation studies should be carried out on multitissue control blocks containing both known-positive and known-negative normal and neoplastic tissues. 369 In veterinary medicine, species-specific tissue microarrays are not commercially available. Species-specific tumor cell lines can be used for IHC controls (eg, for HER-2/neu); 306,310 this approach provides an identical control for comparison of results among laboratories. 291
Peptide controls
The basis behind peptide controls is that formalin-fixed peptide epitopes, covalently attached to glass slides, behave immunochemically like the native protein in tissue sections. 38 Mass production of peptide controls is cheaper than tissue or cell line controls, creates an identical control for numerous assays, and facilitates standardization of IHC protocols.
Reference control tissues for infectious diseases
The presence or absence of an infectious agent in a tissue should be assessed by known IHC reactivity patterns plus another (non-IHC) diagnostic method. 15 For example, Listeria monocytogenes control tissue could be assessed by IHC (immunoreactivity of extracellular or phagocytized bacilli) and by bacterial culture. Similarly, BVD virus control tissue could be assessed by IHC (immunoreactivity in cytoplasm of infected cells) and by virus isolation or PCR. In addition, the presence of characteristic lesions of a particular disease supports use as a standard reference control. 291
IHC protocols for infectious diseases should be species matched with test specimens; alternatively, tissues from other species infected with the same agent can be used (eg, BVD virus identification in test tissue from cervids using a bovine control tissue). Nonspecific binding of primary antibody (species-specific molecular mimicry) or secondary (linker) reagents can occur among species. An example is the use of goat PAbs against Neospora caninum on goat tissue. The anti–goat IgG secondary reagent can bind extensively to endogenous goat IgG in the tissue, resulting in background “noise” that obscures any immunoreactive tachyzoites. 291
Reference controls for neoplastic diseases
Often, the only way to verify the presence of an antigen is by histology, which is not always accurate. Moreover, a negative result for tumor markers does not rule out a particular neoplasm because the marker of interest may not be expressed in a poorly differentiated cell population. Ideally, controls for tumor cell markers should consist of the neoplasm of interest, one with heterogeneous reactivity. 369 Control tissues for IHC tumor markers should also be species matched with test specimens because the detection of an antigen in a particular tumor and animal species does not guarantee similar results in a different species. 291
Negative Tissue Control
A negative tissue control is known not to contain the antigen of interest. 149,369 A negative control should be included in each IHC run to verify the specificity of the primary antibody and the presence or absence of background (false-positive) reactivity; if false-positive reactivity (tissue labeling with a pattern similar or identical to that in the positive control) occurs in the negative control, the test should be considered invalid. 149 At least one ancillary test (eg, PCR, virus isolation) on the same animal should be used to rule out the presence of the antigen of interest. As for the positive control, a negative tissue control should be processed in the same manner as the case material. Similarly, when using multitissue blocks for antibody validation studies or as tissue controls, the specimens must be fixed and processed as for the case material. 369 In practice, only 1 tissue control is used due to the common presence of both positive and negative cells within the positive control. If possible, the positive tissue control section should be on the same slide bearing the diagnostic (test) tissue section. 291
The lack of labeling with a negative tissue control does not necessarily validate labeling in the positive tissue control. An example is observed with some MAbs produced in ascites fluid labeling the Golgi zone of different types of epithelial cells in different species (Fig. 27). 351 This nonspecific labeling is more related to the immunized mouse rather than to the type of immunogen used or the immunoglobulin class (eg, IgG1 or IgG3) produced in the ascites fluid and has been blocked by adsorbing the ascites fluid with blood group A1 human erythrocytes. 351 We have observed similar nonspecific reactivity with some MAbs. In the end, interpretation of specificity is based on the expected location of the antigen in a particular cell or cell compartment. 400
Internal Positive Tissue Controls
Internal positive tissue controls (eg, smooth muscle markers or vimentin in blood vessels) are present in many test tissues. Labeling in these areas indicates appropriate immunoreactivity. In addition, with internal controls, there is no variation in fixation or processing. 32,239 Internal controls can be used to assess sample quality by identifying “housekeeping” or structural proteins present in relatively constant amounts in certain cell types in a wide variety of tissues, such as endothelial cells or lymphocytes. 369
Reagent (Antibody) Controls
Negative reagent controls (without the primary antibody) are used to confirm test specificity and to assess the degree of nonspecific background caused by the secondary or tertiary antibody. 149 Commonly, the primary antibody is replaced by 1 of the following: (1) antibody diluent, (2) same-species nonimmune immunoglobulin at the same concentration and in the same diluent, (3) an irrelevant antibody, or (4) buffer. 306,369 Only reagents 2 and 3 qualify as true-negative controls. 291 Ready-to-use, universal negative reagents are commercially available for rabbit and mouse primary antibodies. With the different quality standards among laboratories, an agreement on which reagent control is best may not be forthcoming. 400 The current CLIS-approved guideline for IHC also accepts, as an inferior option, simple omission of the primary antibody from the protocol. 149
If the diagnostic workup requires a panel of antibodies, each of the same isotype and similar concentration and derived from the same species, the panel of primary antibodies itself can serve as a set of irrelevant reagent controls; thus, the need for multiple negative controls is eliminated. With this method, separate controls should still be run for each different protocol (eg, different AR or detection system). 369
Quality Assurance/Quality Control
Human IHC laboratories must meet federally mandated standards of operation as defined by the Clinical Laboratory Improvement Amendments (CLIA). 101 Veterinary laboratories accredited by the American Association of Veterinary Laboratory Diagnosticians (AAVLD) must meet the ISO/IEC 17025 standards for all types of assays, including IHC. This document establishes the general requirements for testing, including laboratory organization, document control, test calibration, and technical requirements for performing a test. Technical requirements include personnel training and qualifications, environmental conditions, calibration and validation, equipment, and reporting. 168 In nonaccredited laboratories, diagnostic tests should be validated by documentation of internal or interlaboratory performance using reference standards and relevant diagnostic specimens. This should be corroborated by the endorsement of organizations, such as the World Health Organization, by publication in peer-reviewed journals or by direct comparison with an established method. Following are general guidelines for quality control (QC)/quality assurance (QA) standards for veterinary laboratories. More detailed information has been published. 291
Daily Quality Assurance/Quality Control
Each laboratory should establish standard operating procedures (SOPs) for histology. These include schedules for cleaning, maintenance, and monitoring logs, along with a daily check of equipment such as microwave, oven temperature for drying slides, temperature of water baths, pH meter, reagent containers, and so on (Fig. 28). 217 Buffers, antibody dilutions, and other reagents should be prepared according to standard or manufacturers’ recommended procedures.
Internal Quality Assurance/Quality Control
A biannual internal QA/QC check of IHC slide interpretation by pathologists is recommended.
External Quality Assurance/Quality Control
The goal for interlaboratory comparisons is to document variations in reactivity among laboratories and to set minimum QA/QC standards for IHC protocols to be shared by all laboratories. 409 Despite its difficulty, interlaboratory standardization of IHC tests on animal tissues in veterinary laboratories is in progress. Even in human medicine, only a handful of IHC tests (eg, Hercep test) have been validated in such a way that different laboratories can perform tests with consistent results and not without controversy. 28,156,314 As Fletcher and Fletcher 104 pointed out, “Whether we like it or not, the practice of anatomic pathology is to some extent subjective, although we all strive for as much objectivity and reproducibility as possible in our daily work.”
There are 2 main approaches to interlaboratory standardization. 291
Technical equivalency testing of an IHC test
Unstained slides are distributed among different laboratories for performance of the IHC test. Technical equivalency testing evaluates the quality and interlaboratory consistency of IHC and focuses not only on results but also on the technical quality of the IHC preparation and artifacts that might influence the test results (eg, background staining, loss of tissue due to harsh treatment). Currently, the Pathology Committee for the AAVLD Program for Interlaboratory Comparison of Infectious Agent Immunohistochemical Assays recommends an annual, voluntary, immunohistochemical proficiency test managed by the Pathobiology Section of the National Veterinary Laboratory Services, USDA (Ames, IA). Unstained paraffin slides are sent to participating laboratories, which perform the required IHC test and interpret the results. Evaluation includes quality of the IHC procedure and its interpretation. These interlaboratory tests are common in human medicine and have exposed differences in IHC proficiency among laboratories that could affect therapeutic decisions. 68,136,309,380,416,426 For “stand-alone” IHC tests in veterinary medicine (eg, scrapie, chronic wasting disease), participating laboratories must pass an annual proficiency test. 291 Similar requirements have been established for various biomarkers in human tissues. 416,426 External quality assurance for immunocytochemical preparations is limited. 188
Diagnostic equivalency testing using IHC testing as an adjunct
In this case, both the pathologist’s proficiency and the technical quality of IHC are evaluated. A brief history and description of the tissue examined is included with tissue sections. The pathologist must base the diagnosis on routine (eg, hematoxylin and eosin) staining and support it with appropriate IHC testing. Results could be reviewed by a committee to address any differences among participating laboratories. 407
Immunohistochemistry Reporting and Interpretation
If not included in the standard report, the IHC report should contain demographic information pertinent to the case, the tissue that was tested, and the antibody used or the targeted antigen. Other IHC test details should be filed in the laboratory. Results for infectious antigens should be reported as “detected” or “not detected.” The interpretation for infectious disease IHC/immunocytochemistry (ICC) tests should include a disclaimer that any lack of detectable antigen could be due to the absence of the antigen in the section or to technical aspects, such as prolonged fixation or method sensitivity. 291 For tumor markers, a description should include the cellular location (eg, cytoplasmic, cell membrane, nuclear) (Table 6), distribution through the tissue section, labeling intensity, and percent positive cells, along with an interpretation of the test results. 149 If antigen location is atypical, or if testing of a marker has not been validated in the species of interest, this should be stated in the report. 291 A scoring system for IHC has been proposed by CLIA (Tables 7, 8). 149 Personnel who read IHC slides should be familiar with the antibodies used and their specificity and sensitivity, and an experienced pathologist should review daily IHC preparations for QA/QC. 291
Staining Patterns and Manual Interpretive Scoring Criteria.
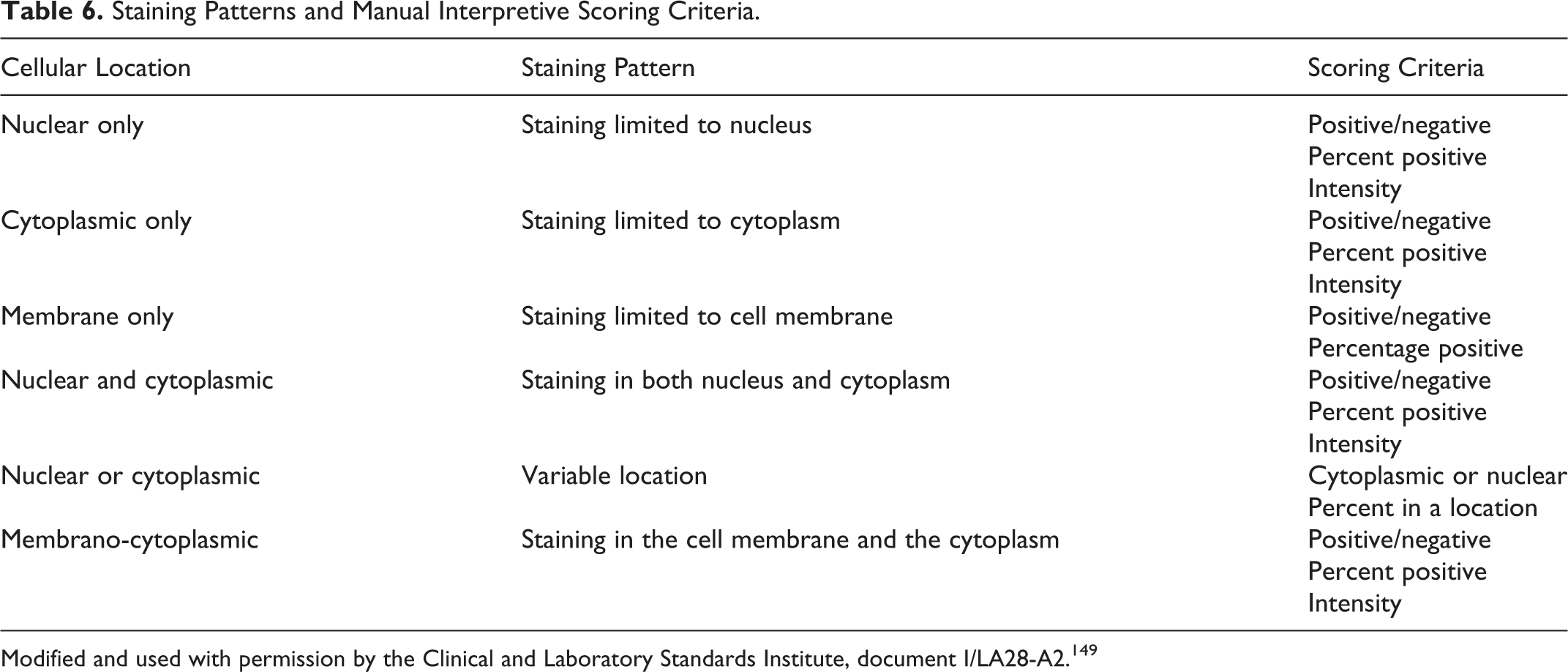
Modified and used with permission by the Clinical and Laboratory Standards Institute, document I/LA28-A2. 149
Immunohistochemical Score Based on Different Percentages of Cytoplasmic Staining.
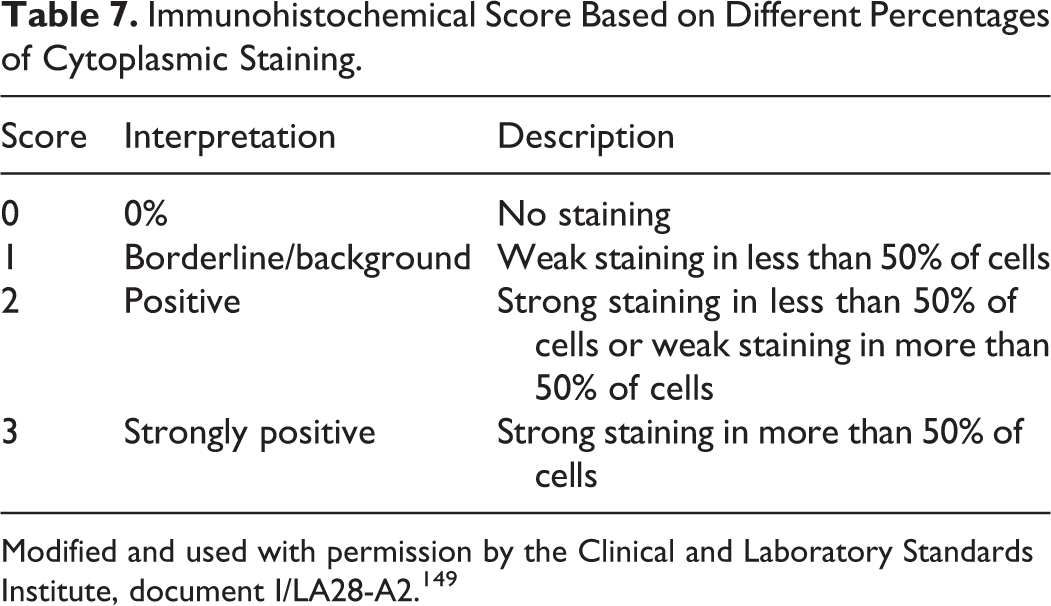
Modified and used with permission by the Clinical and Laboratory Standards Institute, document I/LA28-A2. 149
Scoring Approaches for Immunohistochemistry Nuclear Staining.
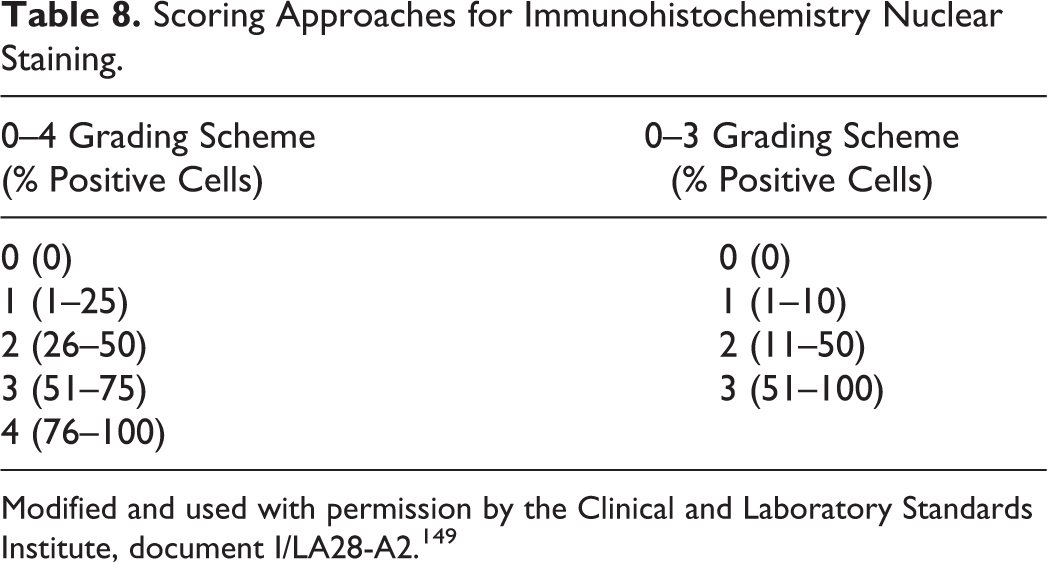
Modified and used with permission by the Clinical and Laboratory Standards Institute, document I/LA28-A2. 149
Most important, interpretation of IHC results requires familiarity with the expected pattern of immunoreactivity based on location of the antigen in the cell of interest. 425 For example, labeling of cytokeratins and vimentin should be cytoplasmic; labeling of TTF1 in lung or thyroid tumors is nuclear. 299,300 The degree of specific immunoreactivity varies among cells and, in some cases, among different cell compartments. Cellular distribution of some antigens has prognostic significance in certain tumors. 191 Finally, interpretation of an IHC reaction is only as good as the controls. 413
Particularly in leukocytic populations, IHC expression of a marker can confirm cell type, without documenting the neoplastic nature of the proliferation. Molecular demonstration of clonality supports a diagnosis of neoplasia, for example, in feline intestinal lymphoma, in which histologic distinction between neoplastic lymphocytes and reactive lymphocytes of inflammatory bowel disease is difficult. 185,190,246
Patterns of Specific Labeling
Antigens can be present in various organelles and intracellular locations or outside cells (Fig. 19). In the nucleus, viral inclusions, inclusions of known or unknown cause, nucleoli, chromatin, the nucleoplasm, and the nuclear membrane may be labeled. Other intracellular sites of antigen expression include mitochondria, lysosomes, rough and smooth endoplasmic reticulum, lipid droplets, peroxisomes, pigment granules, and other organelles or structures. An antigen may be found in several locations, in several different organelles, in both the nucleus and cytoplasm, and in the cytoplasmic membrane and cytoplasm. Outside the cell, connective tissue, tissue matrices, extracellular fluids, and other substances both normal and abnormal can be immunoreactive. Antigens can exist in certain anatomical locations in normal cells but in other locations in neoplastic or reactive cells. Genetic changes (eg, mutations) can result in antigenic shift and redistribution; for instance, β-catenin is typically a cell membrane protein but can be detected in the nucleus in some cases. 27,424 Antigens that exist at low levels or have a short half-life in normal cells (eg, p53) may only be detectable by IHC after mutation.
Patterns of Nonspecific Labeling
Nonspecific background staining can mimic specific immunoreactivity. At the highest antibody concentration, all cells (epithelial, mesenchymal, and neural) and tissues are brown (with DAB as chromogen) (Fig. 20). With increasing dilution, nonspecific staining disappears. Some cell types, especially mast cells and plasma cells, may appear brown at all dilutions depending on the IHC technique, but these cells will also be brown in the negative control slides, which were not exposed to the primary antibody. The careful use of appropriate controls should allow distinction between background and specific labeling.
Immunocytochemistry
ICC is the detection of antigens in cytologic preparations by means of an immunologic reaction visualized by a chemical reaction. 345 More biomarkers are immunoreactive in cytologic preparations than in FFPE tissue sections. 379 There are important methodological differences between IHC and ICC in (1) the type of substrate or sample, (2) storage of unstained samples, (3) fixation, and (4) test validation and controls. 86
Type of Cytologic Preparation
ICC can be performed on cytospins, cell smears, cell blocks, cytoscrape cell blocks, cell cultures, and liquid-based monolayer preparations. 43 , 62 , 86 , 101 , 345 , 435 Cell smears give reproducible results with nuclear markers but are less suitable for cytoplasmic and membrane markers due to the high background produced by cell damage during slide preparation. 345 Cytospin preparations are less susceptible than smears to cell damage. 284 The use of pretreated slides to promote cell adhesion reduces loss of cells during the ICC procedure. 86 Liquid-based cytology using ThinPrep preparations enhances retrieval of cells from small samples with preservation of cellular detail and, theoretically, a reduction of background due to less blood, mucin, and proteinaceous material in the sample. 86 Cell blocks are one of the best choices for ICC, particularly when cells are numerous. 239 , 285 , 286 Cell blocks are processed similarly to surgical pathology specimens, so IHC methods can be used without modifications. 86 In addition, multiple sections can be produced from a single block, so multiple markers can be evaluated in the same cell population. 86 , 286 , 394 However, cell blocks can vary in cellular density and detail, depending on the nature of the lesion, the quality of the fine-needle aspirate, and postprocedural handling of the needle-rinse specimen. 312 Importantly, because cell blocks are fixed in formaldehyde-containing solutions, sections from cell blocks may need to undergo AR procedures. 45 , 339 A more detailed discussion of the pros and cons of cytologic preparation methods is in Fowler and Lachar. 107
ICC can be performed, even repetitively, 252 on smears previously stained with Romanowsky or Papanicolau and in decolorized smears. 1,14,240 However, cell loss, cell disruption (affecting mostly cell membrane and cytoplasmic markers), and antigenic degradation due to repeated passage through graded alcohols can cause suboptimal results. In cases with only 1 slide available and multiple markers to test, the sample is divided into zones and a different antibody is applied to each zone; alternatively, the sample can be divided using tissue transfer techniques. 329 Cell transfer techniques allow the evaluation of multiple markers when few slides are available. 86 If cell transfer from previously stained cytologic smears is anticipated, nonadhesive treated slides should be used. 239
Cytologic Slide Storage
Storage of air-dried preparations for up to 2 weeks at 2°C to 8°C before ICC does not appear to reduce their antigenicity. 100 If longer storage is needed, slides should be kept at –70°C. 345,362 Some laboratories fix cytologic samples with methanol and, if not used immediately, cover them with 3% polyethylene glycol for storage or transportation. 188
Fixation and Antigen Retrieval
Another difference between IHC and ICC is the type of fixation. In contrast to IHC, in which 10% formalin is used routinely, there is no standard fixative for cytologic specimens. In general, the type of fixation is determined by the cytological procedure and the antigens to be tested. 345 In ICC, samples are wet-fixed or air-dried and fixed immediately before performing ICC with 100% acetone or 10% formalin. 73,379 Air-dried preparations tend to lose fewer cells than wet-fixed preparations, but inconsistent ICC results have been reported with air-dried samples. 86 For nuclear antigens, fixation of air-dried specimens in buffered 4% to 10% formalin alone or followed by methanol-acetone produces excellent results, 345,361 although air-drying of smears before fixation has been reported to hinder estrogen or progesterone receptor detection. 239 For membrane and cytoplasmic antigens, the type of fixation is not as critical as for nuclear antigens; various fixatives, including formalin followed by ethanol, a 1:1 mixture of methanol-absolute ethanol, or acetone at –20°C, produce good results. 345 However, acetone solubilizes cell membranes, leading to diffusion of small peptides out of cells and false-negative results (Fig. 6). 383 Antigens such as S100 protein, Hep Par 1, and gross cystic fluid protein 15 are leached by alcohol fixatives, producing false-negative results. 62 One laboratory proposes these guidelines: samples should be fixed immediately prior to the ICC procedure; for lymphoid and melanoma markers, samples should be fixed for 5 to 10 minutes at RT in acetone; for epithelial markers, 5 minutes at RT in 95% ethanol or 1:1 mixture of methanol and 100% ethanol; and for nuclear antigens, 3.7% buffered formalin for 15 minutes. 100 Formal saline has been proposed as a “universal” ICC fixative. 207 Others advocate air-drying slides if a lymphoma is suspected and immediately fixing smears in 95% ethanol (without air-drying) in all other cases. 239 A recent comparison of FFPE cell blocks (CBs) with alcohol-fixed centrifuged preparations (AFCPs) did not find significant differences. 166 Cytologic preparations fixed in acetone may be more susceptible to cell loss than those fixed in formalin when using automatic stainers because of weaker cytoplasmic attachment to the glass slides.
AFCPs without AR performed similarly to cell blocks with AR in many cases; however, for some tests, particularly to detect nuclear antigens, AR has improved the reactivity of AFCPs. 166 HIER using citrate buffer pH 6.0 is necessary for most nuclear antigens on ethanol-fixed cytologic smears; for some cytoplasmic membrane and cytoplasmic antigens, it enhances the immunoreaction. 80 Few cytoplasmic membrane antigens require HIER with higher pH buffers. 80 Some laboratories perform HIER irrespective of the fixation type. 435 Enzymatic AR is much less commonly used than HIER in ICC. 435 The mechanism of action of HIER might depend on the fixative. When the amount of cytologic specimen is limited, the same slide can be tested for a second marker if the first test is negative. 74 Adhesive slides should be used to perform HIER or protease AR on smears; otherwise, the sample is likely to detach. 239
Immunocytochemical Method
Immunocytochemical procedures are similar to those for IHC. 286 Endogenous peroxidase is blocked with 3% hydrogen peroxide; this step is unnecessary in acetone-fixed samples. In blood-rich smears, the use of alkaline phosphatase procedures rather than immunoperoxidase avoids the background produced by endogenous peroxidase without the need for strong quenching reagents. 86 Antigen retrieval is often necessary even without formalin fixation. Unfortunately, the wide range of fixation procedures makes interlaboratory standardization of AR difficult; each laboratory must optimize the procedure for each antigen. 188,286,341 Remarkably, in a multi-institutional study, IHC quality was good irrespective of the cytologic preparation or fixation procedure, except that cell block preparations consistently had the highest scores. 188
Controls and Standardization in Immunocytochemistry
Other challenges in ICC are (1) use of adequate positive and negative controls (ideally, controls should be treated in the same way as test samples), (2) nonstandardized methodology and antibody titers, (3) difficulty in evaluation of immunoreactivity in inflamed or necrotic samples, (4) difficulty in distinguishing normal from neoplastic cells, and (5) deleterious effect of protease digestion in non–formalin-fixed material. 107,188
Positive and negative controls are standardized in human and veterinary IHC 284,286 and should be included with each test sample in ICC. 62 Although the ideal tissue control is a comparably fixed cytologic preparation, 62 only 13% of publications listed positive and negative controls processed identically as the samples; 54% did not mention the use of controls or processed controls separately. 64 The College of American Pathologists recognizes the impracticality of maintaining separate positive control samples for every possible combination of fixation, processing, and specimen type (see comment for questions ANP 22550 in the Anatomic Pathology checklist at http://www.cap.org/apps/cap.portal). Cytologic control preparations fixed in acetone lose antigenicity after several months even if wrapped in aluminum foil and refrigerated; control samples fixed in formalin retain their antigenicity indefinitely. 379 The production of cytologic controls from organs (eg, lymph node, liver) is described elsewhere. 379
Interpretation
As in IHC, the interpretation of ICC requires consideration of the “antibody personality profile” and the “infidelity” (cross-reaction with antigens in other cell types) of tumor-specific markers. 425 First, a positive or negative reaction must be defined. 286 With the general lack of clear guidelines, each laboratory should document and state in the ICC report what is interpreted as a positive result. Alternatively, as recommended in human IHC, 124 a statement in the ICC report indicating the intensity of the labeling and percentage of positive cells (of the type of interest) is more informative than just a positive versus negative result. As for IHC, interpretation of ICC tests should be done in conjunction with a standard cytologic stain (eg, Wright-Giemsa, Papanicolaou) and in concert with the clinicopathologic correlations. 62
Causes of false-positive results in ICC include inadequate fixation, insufficient blocking of endogenous peroxidase or biotin activities, and high background labeling with PAbs, or detection of immunoreactivity in the wrong cell type (eg, normal vs neoplastic). 62,239,345 Two other causes of false-positive results are spurious staining of cells that have phagocytized other cells and reagent trapping in clusters or layers of cells. 239 False-negative results are caused by improper fixation, inadequate antibody titration, insufficient AR, or cell damage during preparation. 62,345
The Future of Immunohistochemistry
Since IHC was developed in the 1940s, 65 major advances have been made in the sensitivity and specificity of the method and the availability of biomarkers for FFPE tissues. The main purpose of IHC has been to characterize a neoplasm or identify an infectious agent. However, the current trend is to apply IHC to the detection of genetic abnormalities in neoplasms, detect findings of prognostic importance, and aid in selection of agents for cancer treatment. 208 These topics are actively investigated in human medicine and not without controversy. Before applying IHC to such investigations, the entire process must be standardized, from the time a sample is taken from the patient (preanalytical variables) through the analytical steps and ending with the IHC results and diagnosis (postanalytical variables). || Standardization of IHC procedures will be necessary to compare results among laboratories and will be mandatory if computer-assisted quantification is intended, particularly if those results influence the therapeutic approach. Although the barriers to progress are daunting, the in situ detection of protein expression by IHC will be a vital part of pathology for years to come, even if the generation, interpretation, and application of test results in the context of disease change drastically.
Footnotes
Acknowledgements
We thank Marilyn Beissenherz, VMDL–University of Missouri, and Dee DuSold, ADDL–Purdue University, for their dedication to preparing IHC slides and troubleshooting. We also thank Dr Chang Kim, Comparative Pathobiology, Purdue University, West Lafayette, Indiana, and Dr van der Loos, Academic Medical Center, Amsterdam, the Netherlands, for reviewing portions of this manuscript. Finally, we thank the veterinarians who submitted cases for IHC to the laboratories in which the authors have worked and learned the red, brown, and blue technique.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.