
Abstract
Immunohistochemistry is an integral technique in many veterinary laboratories for diagnostic and research purposes. In the last decade, the ability to detect antigens (Ags) in tissue sections has improved dramatically, mainly by countering the deleterious effects of formaldehyde with antigen retrieval (AR) and increasing sensitivity of the detection systems. In this review, I address these topics and provide an overview of technical aspects of immunohistochemistry, including those related to antibodies (Abs) and Ags, fixation, AR, detection methods, background, and troubleshooting. Microarray technology and the use of rabbit monoclonal Abs in immunohistochemistry are also discussed.
The publication of a paper by Coons et al. in 1941 23 describing an immunofluorescence technique for detecting cellular antigens in tissue sections marked the beginning of immunohistochemistry (IHC). Since then, IHC has become a valuable tool in both diagnosis and research of infectious and neoplastic diseases in a variety of animals. The basis of IHC is very simple and bridges three scientific disciplines: immunology, histology, and chemistry. The fundamental concept behind IHC is the demonstration of antigens (Ag) within tissue sections by means of specific antibodies (Abs). Once antigen–antibody (Ag-Ab) binding occurs, it is demonstrated with a colored histochemical reaction visible by light microscopy or fluorochromes with ultraviolet light. Although conceptually simple, the methodology of IHC has become more complex as more stringent goals of sensitivity and specificity are established (Fig. 1). 91 Initially, simple (direct) methods were used that produced quick results but lacked sensitivity. Currently, extremely sensitive methods are available to detect one or multiple Ags simultaneously or even to examine hundreds of tissues in the same section for the presence of a particular Ag (microarray technology). Another critical advance in the 1990s was techniques to retrieve Ags that had been altered by fixation by means of heat, increasing exponentially the number of Ags detectable in routinely fixed tissues. Veterinary pathologists face many challenges when performing IHC because of the diversity of species studied and no guarantees that Abs will cross-react among different species. IHC has been established as a solid and reliable methodology for both routine diagnostic and research activities in veterinary medicine. This review will cover technical aspects of immuno-histochemistry, including those related to Abs and Ags, fixation, AR, detection methods, background, and troubleshooting. Because IHC can appear intimidating, one of the goals of this review is to help all readers understand technical aspects of IHC and keep them from fearing this “red, brown, and blue technique.”
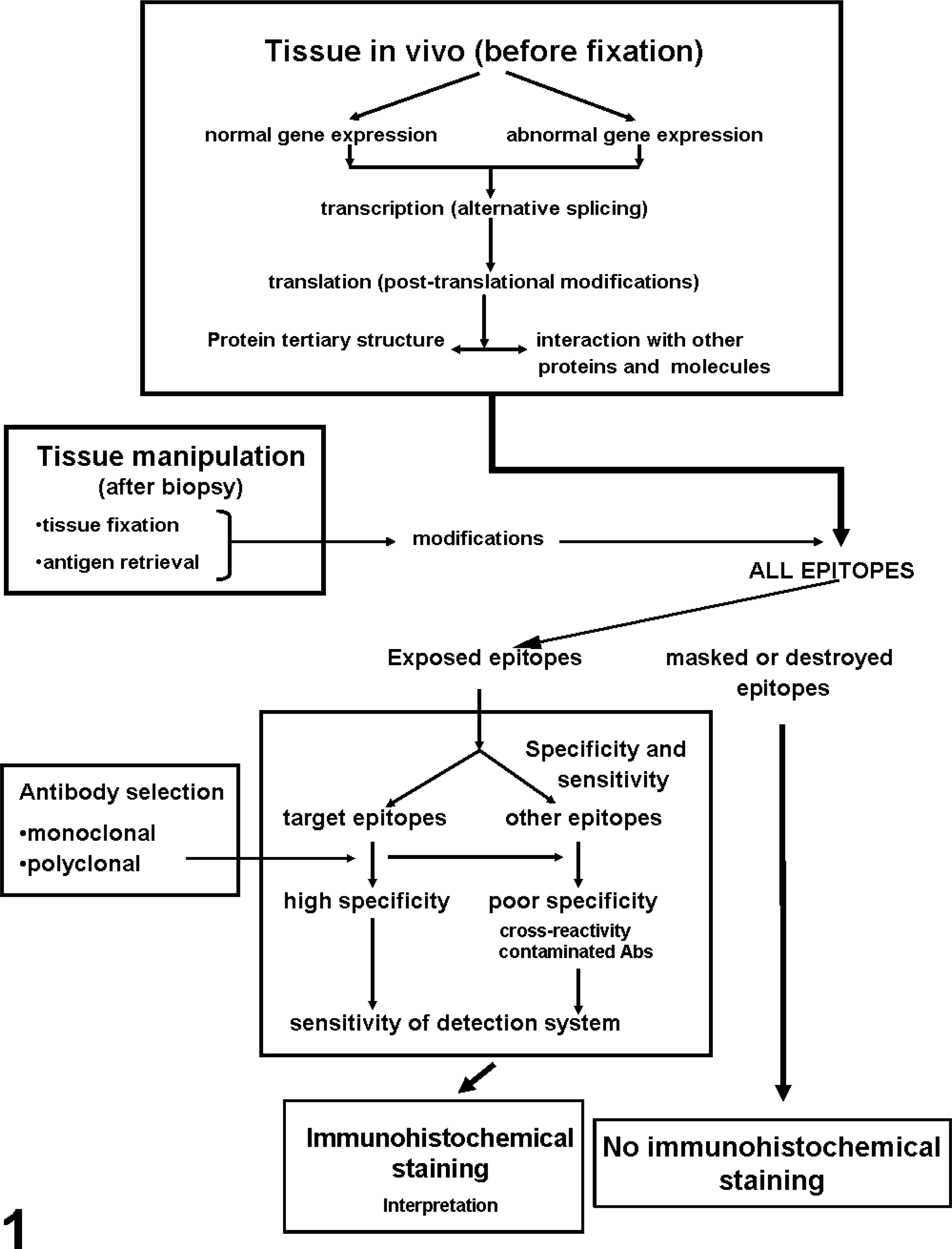
Principal factors affecting the outcome of immunohistochemical studies (reprinted from Mighell et al. 91 with permission from Blackwell Publishers).
Antigens and Antibodies
Immunohistochemistry is based on the binding of Abs to a specific Ag in tissue sections. The most common immunoglobulin (Ig) used in immunohistochemistry is IgG; IgM is less commonly used.
Antibody structure
Igs are Y-shaped and consist of two identical light chains and two identical heavy chains (Fig. 2). The heavy chains determine the Ab class. The tail of the Y is called Fc. The light chains of most vertebrates have two distinct forms, called kappa and lambda. In any Ig molecule, both light chains and both heavy chains are of the same type. The light chains consist of two distinct regions: the C-terminal half of the chain is constant and called CL (constant: light chain), whereas the N-terminal half of the chain has abundant sequence variability and is called the VL (variable: light chain) region. The Fab region is the Ag-binding portion of the Ig and contains variable and constant segments of the heavy and light chains. The Fc portion of the Ab determines the biological functions and permits Ab binding to other Ab, complement, and inflammatory cells with Fc receptors. This portion of the Ig is needed for multistep immunohistochemical techniques. This part is also responsible for background staining resulting from nonimmune adherence of Abs to the tissue section; to avoid that, there has been a trend to use only Fab or F(ab)2 portions of the Ig molecule for IHC. The major problem with this approach is that the Fc portion of Igs tends to stabilize Ab binding to solid substrates such as tissue. The specific binding of an Ab to an Ag occurs via hypervariable regions of both heavy and light chains of the amino terminus. The Ag binding site of an Ab is called the paratope. Epitopes are the regions of an Ag that bind to Abs. 49 Epitopes are usually 5–21 amino acids long. 118 One of the most important criteria for binding of an Ab is the tertiary structure of the epitope, or the way in which the peptide chains of a protein are folded together or interact with adjacent peptides. The paratope interacts with the tertiary structure of the epitope through a series of noncovalent bonding interactions (see below). The more bonding interactions, the greater the affinity and avidity (defined as the overall binding strength between the Ag and Ab) of the Ab. IgG Abs are bivalent (have two identical arms used in Ag recognition). This is a key feature necessary to perform multiple-layer immunohistochemical methods.
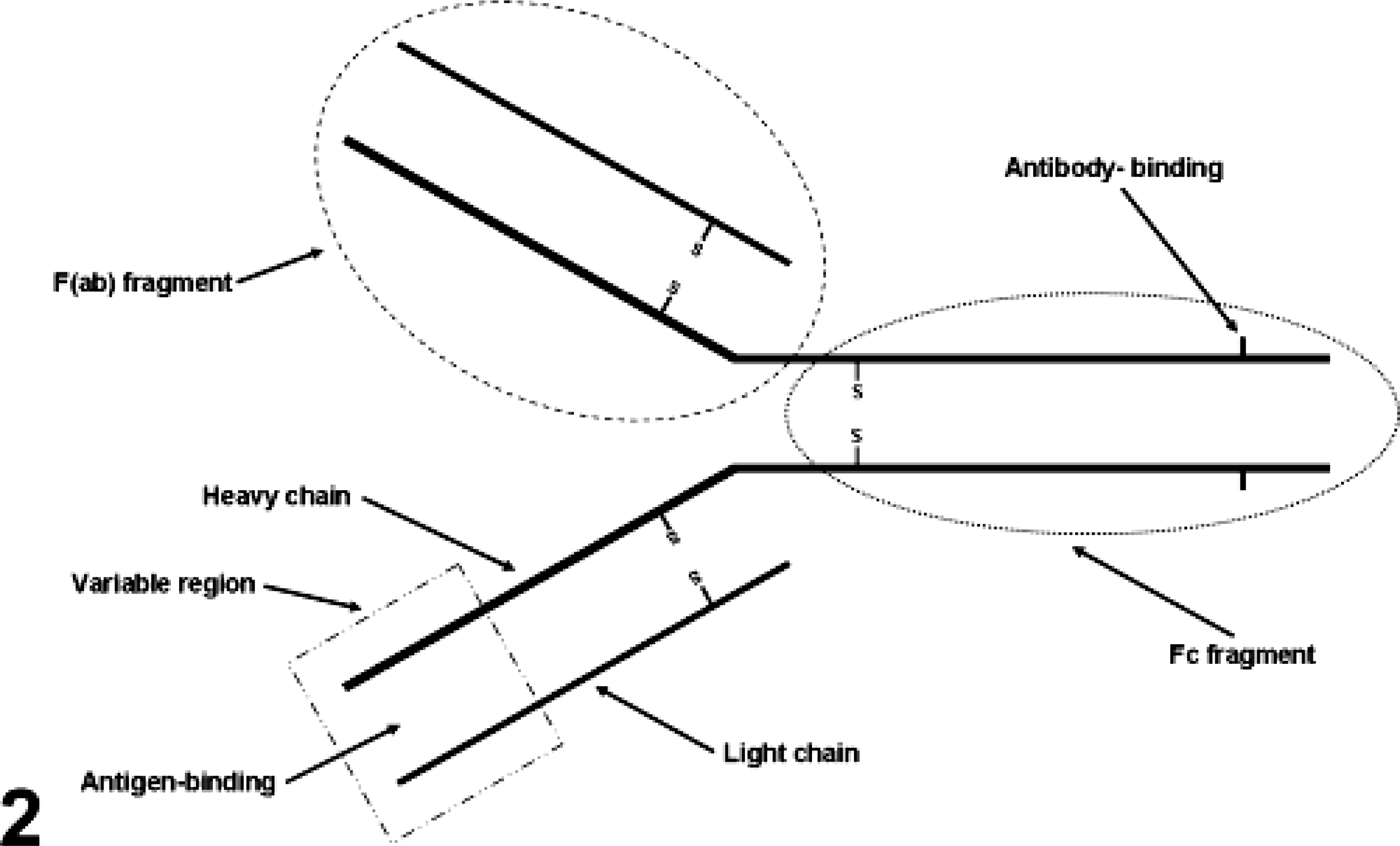
Structure of an Ig. The area delimited by an oval dashed line corresponds to the F(ab) fragment. The area delimited by a square is the variable region. The area limited by an oval shaped dotted line is the Fc fragment.
The nature of Ag-Ab interactions
From a chemical and biochemical point of view, Ag-Ab interactions are somewhat unusual. The bonds involved are weak (mostly hydrophobic and electrostatic) and not covalent. Hydrophobic bonds happen between macromolecules with surface tensions lower than that of water. They can be interatomic or intermolecular. These hydrophobic interactions are imparted primarily through the side chain amino acids phenylalanine, tyrosine, and tryptophan. 11 By their lower attraction to water molecules, these amino acids tend to link to one another. Electrostatic (Coulombic) interactions are caused by attractive forces between one or more ionized sides of the Ag determinant and oppositely charged ions on the Ab-active site. These typically are the carboxyl and the amino groups of polar amino acids of the Ag and Ab molecules. Van der Waals forces are weak electrostatic interactions between dipolar molecules or atoms. 1 Van der Waals forces and electrostatic attractions are maximal at the shortest distances. Therefore, precise juxtaposition of oppositely charged ions on epitopes and paratopes favors strong electrostatic bonding. 1 Hydrogen bonds are the result of dipole interactions between OH and C=O, NH and C=O, and NH and OH groups. The binding energy is of the same order of magnitude as that of van der Waals and electrostatic interactions. Its significance in Ag-Ab interactions is probably not great because of the necessity of a very precise fit between both molecules for it to happen. Although there are cases in which only one of these types of interactions is significant in Ag-Ab binding, for most polysaccharides, glycoproteins, and polypeptide Ags, the Ag-Ab bond is a combination of van der Waals forces and electrostatic interactions. 1 Abs, except for IgM (which is dekavalent), are divalent. Most protein Ags are multivalent. Each valency site of protein Ags generally is an antigenic determinant (epitope) with a completely different configuration from all the other valency sites (e.g., a monoclonal Ab can react with only one valency site of such a protein Ag). 152 Table 1 indicates conditions favoring Ag-Ab binding and association.
Main forces involved in antigen–antibody binding. 152

Affinity is a thermodynamic expression of the binding energy of an Ab (paratope) to an antigenic determinant (epitope). Affinity can be defined in mathematical terms as an affinity constant (KA), which represents the amount of Ag-Ab complex that will be formed at equilibrium.
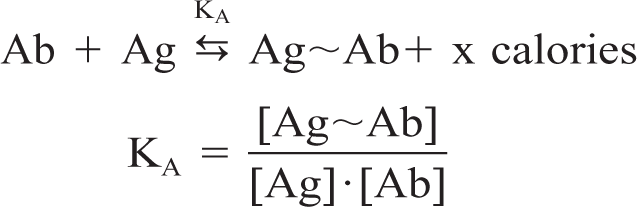
where Ag∼Ab are Ag-Ab complexes and the terms in square brackets indicate molar concentrations. The range of Ag-Ab affinity constants is wide and varies from below 105 liters/mol to 1012 liters/mol. What do these numbers mean? An Ag-Ab complex with a KA of 1012 liters/mol has a 1,000-fold greater affinity than an Ag-Ab complex with a KA of 109 liters/mol. The affinity of the Ag-Ab reaction is important for practical reasons: 1) high-affinity Abs will bind more Ag in a shorter incubation time than low-affinity Abs, and 2) in general, the higher the affinity the more dilute the Ab solution can be. 40
Ags
Ags can have different structures (isoforms). A single gene can generate several different protein isoforms via two principal mechanisms. 1) Alternative splicing of the primary gene transcript can produce multiple different mature transcripts, each of which codes for a slightly different protein. 125 Many proteins undergo posttranslational modifications, such as glycosilation, phosphorylation, and proteolytic processing, which add more complexity to proteins derived from a single gene (Fig. 1). 91 As a result of these mechanisms, a single gene can generate numerous protein isoforms and even this repertoire can change with time (e.g., tenascin and hemoglobin isoforms that change from fetal development to adulthood). 91
Selection of immunogens
The source and preparation of the immunogen to stimulate production of Abs is very important to obtain the best quality of reagent for IHC. Two broad groups of immunogens exist: synthetic peptides and purified protein preparations. 91 Synthetic peptides have the advantage of a known amino acid sequence. This information might be essential in the interpretation of IHC studies, both with respect to the isoforms of the target protein that can be detected and any cross-reactivity with similar peptide sequences in other proteins. Synthetic peptides also have potential disadvantages. An isolated synthetic peptide sequence might lack the normal three-dimensional structure of the native protein. In addition, other proteins can be intimately associated with the protein of interest in vivo. Both of these factors could mask the target epitopes, prevent detection in vivo by Abs raised to synthetic peptides, and therefore yield false negative results. A third disadvantage is that in vivo posttranslational modifications can be crucial in the final Ag product, and they are not present in synthetic peptides. 81, 82 Use of purified proteins as immunogens avoids many of the problems generated by the use of synthetic peptides. 91 However, purification of a protein to homogeneity from either cells or tissues can be technically difficult. Also, contaminating proteins might be more antigenic than the protein of interest, producing a disproportionate and unwanted immunogenic response. Another problem when using purified proteins is when the targeted Ag includes highly immunogenic epitopes that are not specific to the Ag of interest. This is the typical situation with very similar posttranslational modifications among Ags that otherwise are very different. The source and details of Ag purification, as well as the isoforms of the targeted protein recognized by an Ab preparation, are seldom reported for commercially available primary Abs, limiting interpretation of patterns of staining in IHC studies. 91 Other factors affecting immunogenicity include size, conformation, and electrical charge of the Ag; the use of adjuvants; dose of the Ag; and route of administration.
Monoclonal and polyclonal Abs
Abs are made by immunizing animals (mouse, rabbit, goat, horse, etc.) with purified Ag. The animal responds by producing Abs that specifically recognize and bind to the Ag. Polyclonal Abs are produced in multiple animal species, particularly rabbit, horse, goat, and chicken. Polyclonal Abs have higher affinity and wide reactivity but lower specificity when compared with monoclonal Abs. 49 Polyclonal antisera include not only several different Abs to the target protein, but irrelevant Abs can also be present in high concentration (up to 10 mg/ml) if not affinity purified. 31, 91 Polyclonal Abs have the advantage over monoclonal antibodies in that they are more likely to identify multiple isoforms (epitopes) of the target protein. However, a polyclonal preparation could be very heterogeneous because of the presence of Abs to strong and weakly immunogenic epitopes in the same preparation. 91 Variations in Ab titer and quality, depending on the animal immunized, also contribute to variance among batches of Abs. 98 The “immunologic promiscuity” of polyclonal Abs that can be an advantage (e.g., more possibilities of detecting an Ag through multiplicity of epitope recognition) can also be a disadvantage; the greater the number of different Abs to the target protein in a single preparation, the greater the likehood of cross-reactivity with similar epitopes in other proteins and, therefore, the likelihood of false positives. 91 For instance, common immuno-genic domains of fibronectin III are present in at least 65 unrelated proteins. 14 Monoclonal Abs. Monoclonal Abs are produced mostly in mice. Köhler and Milstein 73 developed the technology of monoclonal Ab production. Mice are injected with purified immunogen (Ag). After an immune response has been achieved, the B lymphocytes (Ab-producing cells) are harvested from the spleen. Because isolated B cells have a limited life span, they are fused with mouse myeloma cells. This is followed by selection of hybridomas of desired specificity. The hybrid cell produced (hybridoma) is an immortal cell that produces Igs specific for a single epitope (monoclonal Abs). The advantage of monoclonal Abs is their higher specificity when compared with polyclonal Abs (Table 2). Hybridomas can be maintained in cell cultures (highly pure Ab but at low concentration) or in the peritoneum of mice (ascitic fluid, which has a 10–100-fold higher concentration of Ab than in cell culture but has nonspecific proteins and endogenous Igs from mice). This specificity reduces (although does not remove completely) the possibility of cross-reactivity with other Ags. One reason for cross-reactivity is that monoclonal Abs are directed against epitopes consisting of a small number of amino acids, which can be part of several types of proteins and peptides. 97 For instance, an anti-human proinsulin Ab cross-reacts with both insulin and glucagon-secreting cells. 9 Because this binding is to an identical epitope, the staining characteristics are almost or completely identical to the intended Ag epitope and, therefore, very difficult to distinguish. 143 In some instances, it is difficult to determine whether the immunoreactivity is owing to shared epitopes (cross-reactivity) or epitopes resulting from protein cross-linking during fixation with aldehydes. 49, 68 Background staining of monoclonal Abs because of nonspecific Igs is reduced (ascites fluid) or nonexistent (cell culture supernatant). Rabbit monoclonal Abs also are commercially available. Their production has been facilitated by the development of Ab libraries. 112, 114 The advantage of rabbit monoclonals over mouse monoclonals is their higher affinity, suitability for use on mouse tissues without special procedures, increased specificity in some cases, and avoidance of AR methods. 20, 46, 78
Advantages of monoclonal and polyclonal antibodies.
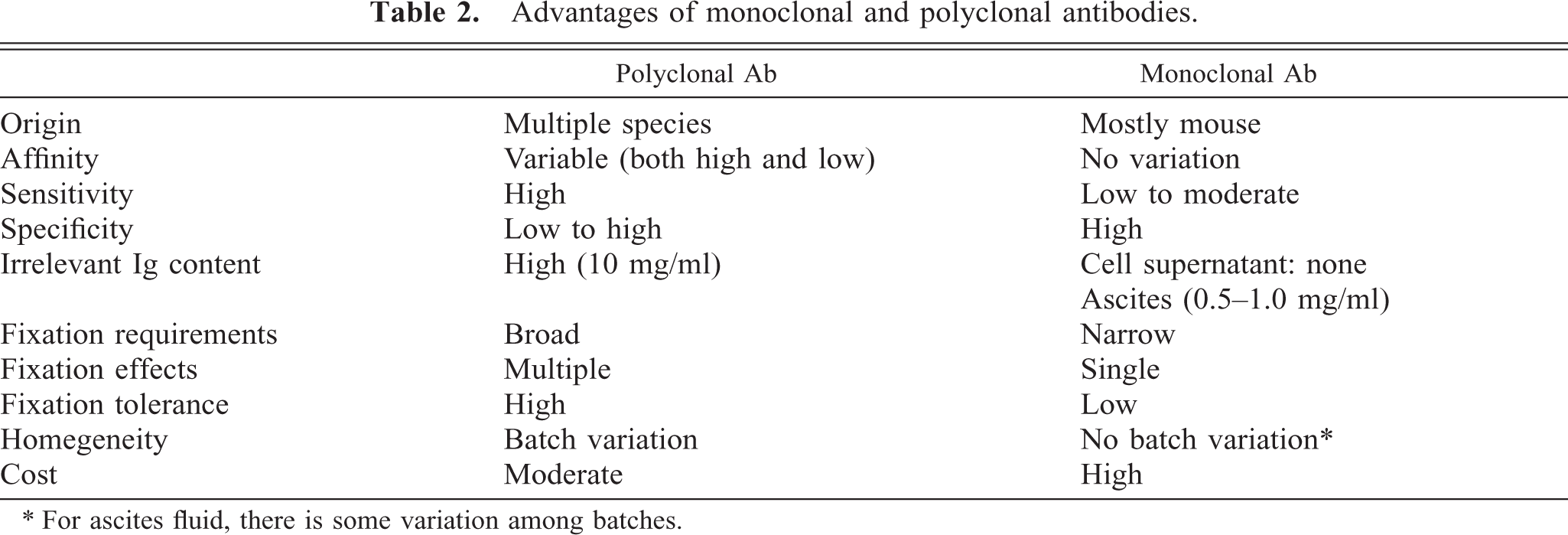
∗ For ascites fluid, there is some variation among batches.
Presentation of commercial Abs
The amount of information in commercial catalogs about Abs targeting the same Ag varies a lot depending on the manufacturer. It is becoming more common, particularly with companies specialized in the production of antisera, to include the following information: format of the Ab (e.g., purified, whole serum, supernatant, ascites, Ig isotype), host in which the Ab was produced (e.g., mouse, rabbit), protein concentration, immunogen used (including epitope and molecular mass, if known), species reactivity (e.g., human, mouse, others not known), cellular localization (e.g., cytoplasmic, membrane, nuclear), positive tissue control recommended, application (e.g., immunoprecipitation, western blotting, enzyme-linked immunosorbent assay, immunohistology–formalin/paraffin, and frozen), and pertinent literature. Many companies also have images of the immunoreaction in their catalogs, as well as gels with the molecular mass of the epitope(s) detected by the Ab. It is advisable to contact the manufacturer for additional information pertaining to reactivity under certain conditions (formalin fixation) or species cross-reactivity not indicated in the catalog. Other data not commonly available from manufacturers of antisera are the affinity constant of Abs and the possibility of cross-reactivities with other Ags (e.g., serotypes of viruses, other related viruses or bacteria, cell Ags) that might be a result of fixation or AR.
Tissue Microarrays
Tissue microarrays were introduced by Kononen et al. 74 The tissue microarray technology allows simultaneous examination of hundreds of samples on a single microscope slide. This technology has been used not only for detection of proteins in tumor cells, but also for the expression of genes. 53, 65, 93, 99, 100, 103, 120, 134, 147 Tissue microarray technologies include multitumor microarrays (samples from multiple histological tumor types), progression microarrays (samples of different stages of tumor progression within a given organ), prognosis microarray (samples from which clinical follow-up data are available), and cryomicroarrays (frozen samples that might be more suitable than formalin-fixed tissues for detection of RNA). 99, 120, 145 The advantages of the tissue microarray method are: savings in reagents, reduction of technical time, reduction in results variability, feasibility of digitizing immunostain results, and interpretation of results by creating hierarchical cluster analysis. 65, 100, 117 Tissue microarray has the advantage over DNA microarray, in that the cell expressing a particular gene can be identified, whereas in DNA microarray, the sample is digested before being tested; therefore, cell expression cannot be characterized. 43 Although the concept of tissue microarray is simple, this method also has some disadvantages when compared with the classic single sample per microscope slide: it requires highly technical skills to prepare the array and careful planning. Also, selection of the sample is critical because of its small size and usual heterogeneity. 64, 65, 103
A novel technology, the multiplex immunostaining chip (MI chip), is intended to examine multiple Ags in the same tissue section. MI chip technology differs from tissue microarray in that MI permits the analysis of expression of as many as 50 Ags in a single specimen, whereas the microarray technology permits the analysis of a single Ag in many specimens simultaneously. 42 The main problem in clinical application of this technology is the heterogeneity of most tumor sections and, therefore, the possibility of false-negative results. 42
Fixation
Fixation of tissues is necessary to 1) adequately preserve cellular components, including soluble and structural proteins; 2) prevent autolysis and displacement of cell constituents, including Ags and enzymes; 3) stabilize cellular materials against deleterious effects of subsequent procedures; and 4) facilitate conventional staining and immunostaining. 51 Two types of fixatives are used in histopathology: cross-linking (non-coagulating) fixatives and coagulating fixatives.
Formaldehyde and cross-linking fixatives
Formaldehyde is the gold standard of fixatives for routine histology and immunohistochemistry. Formaldehyde preserves mainly peptides and the general structure of cellular organelles. It also interacts with nucleic acids but has little effect on carbohydrates. 34 It is a good preservative of lipids if the fixative contains calcium. 67 In solution, formaldehyde is capable of binding the following amino acids: lysine, tyrosine, asparagines, histidine, arginine, cysteine, and glutamine. 129 The basic mechanism of fixation with formaldehyde is the formation of addition products between the formalin and uncharged reactive amino groups (–NH or NH2), forming cross-links. 26 Once the addition product (reactive hydroxy methyl compound) is formed, additional cross-linking will happen. Thus, in the presence of a second reactive hydrogen, the hydroxymethyl group will form a methylene bridge (Fig. 4).
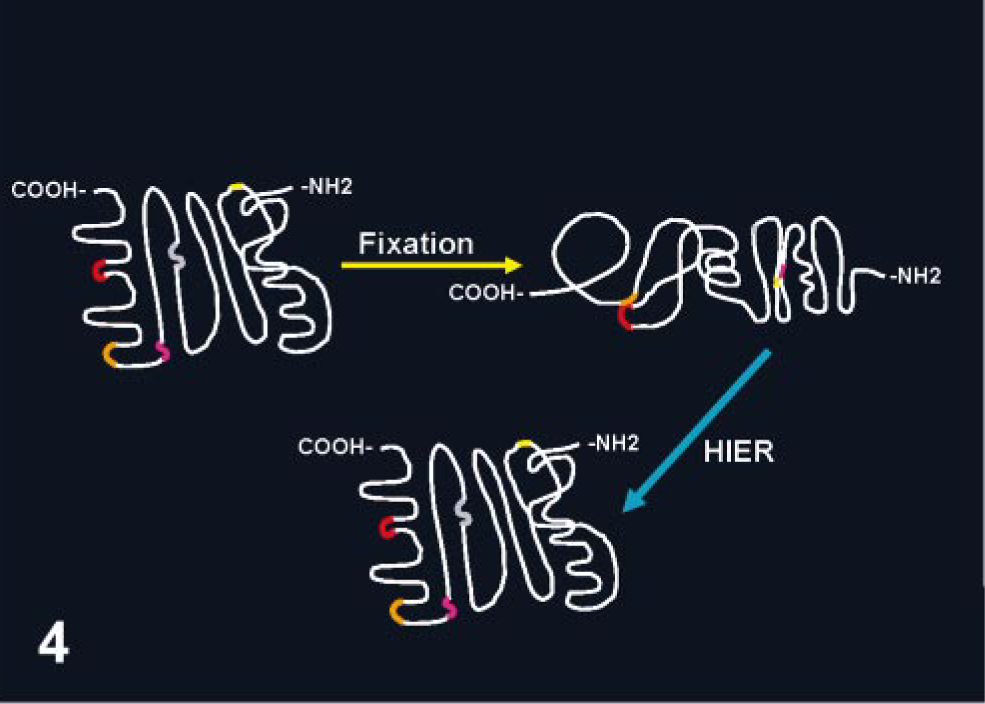
Conformational changes in the protein structure after fixation with cross-linking fixatives. The presentation of different epitopes (small segments in different colors) is modified, which might preclude access to specific antibodies. Heat-based AR methods (HIER) apparently reverse conformational changes produced by the fixative.
Formation of addition products
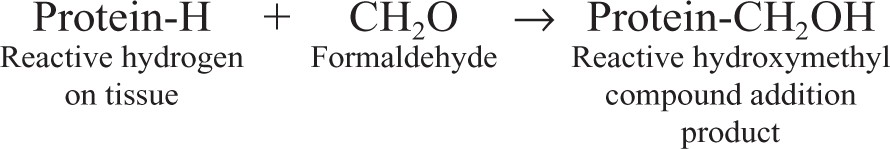
Formation of methylene bridges
The final result of formaldehyde fixation is a profound change in the conformation of macromolecules, which could make the recognition of proteins (Ags) by Abs impossible or, at best, difficult. 94 These changes modify the three-dimensional (tertiary and quaternary) structure of proteins, whereas the primary and secondary structures are little affected. 51, 88 Formalin fixation is a progressive, time- and temperature-dependent process. Overfixation can produce false negative results in IHC from excessive cross-links. 51 However, under-fixation can also produce unexpected results. If large samples are fixed for only 24–48 hours or small biopsies for only several hours, cross-links will happen only in the periphery of the specimen, with the core of the tissue block unfixed or fixed only by coagulation with the alcohol used for dehydration during tissue processing. 51 The effects of formaldehyde can be partially reversed, 34 but glutaraldehyde fixation is considered irreversible. 33 Prolonged washing of fixed tissues will reduce further fixation by removing unbound formaldehyde, 51 although cross-links that already have occurred will remain. 34 Long-term storage of formalin-fixed tissues in alcohol will stop the formation of additional cross-links and, therefore, will have a beneficial effect in Ag detection if these tissues are needed eventually for immunohistochemistry. Overfixation can also be partially corrected by soaking tissue in concentrated ammonia plus 20% chloral hydrate. 76
The pH of a fixative buffer dramatically influences the degree of cross-links. 34, 40 Amino acids are amphoteric substances (contain both acid, −COOH, and basic, −NH2, groups); therefore, they are influenced by pH. Amino acids are charged (–NH3 +) at a lower pH (acid) and uncharged (–NH2) at a higher pH. When using neutral buffered formalin, the pH is shifted to neutrality, causing dissociation of hydrogen ions from the charged amino groups (–NH3 +) of the side chains of proteins, resulting in uncharged amino groups (–NH2). These uncharged groups contain reactive hydrogen that can react with formalin to form addition groups and cross-links. In other words, the use of 10% buffered formalin will produce more cross-links than non-buffered formalin and therefore will have more deleterious effects for immunohistochemistry. 51 If acid formalin is used as the fixative, there is less cross-linking; therefore, the AR procedure will need some modifications. Formalin also disrupts hydrogen bonding and electrostatic interactions between amino acids and other molecules. This results in the disruption of secondary and tertiary structures of proteins, which causes them to precipitate. 40
There is no optimal standard fixation time for every Ag. The chemical structure of the Ag, as well as its relation to other proteins, most likely influences the effect of fixation on the immunoreactivity of Ags. Although in general overfixation is deleterious for the immunoreactivity of Ags, underfixation (e.g., <24 hours) can produce suboptimal results in some instances. 63 The effect of prolonged fixation with formaldehyde on an Ag located in different cell compartments can vary. 51 An example is the irreversible loss of immunoreactivity for Bcl-2 in the nucleus after prolonged formalin fixation, even after heat AR, whereas its cytoplasmic immunoreactivity is preserved or increased after AR. 56 The duration of fixation can alter immunohistochemical reactions, resulting in failure to detect an Ag, weak reaction, increased background, detection of the Ag in an unexpected cell compartment, or altered cross-reactivity.
Other fixatives
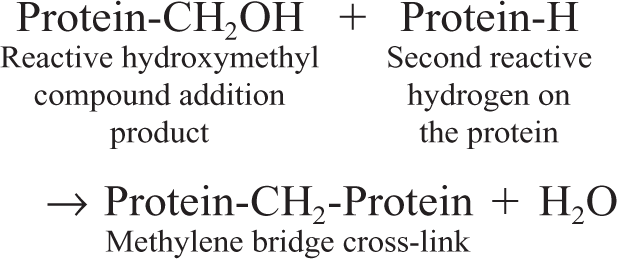
The problems generated by formaldehyde (particularly before the development of AR methods with heat), specifically, the loss of immunoreactivity, have prompted researchers to find alternatives to this fixative. Many of the formalin substitutes are coagulating fixatives that precipitate proteins by breaking hydrogen bonds in the absence of protein cross-linking. The typical non–cross-linking fixative is ethanol. Most proteins in body fluids have their protein hydrophilic moieties in contact with water and hydrophobic moieties in closer contact with each other, stabilizing hydrophobic bonding. 34 Removing water with ethanol destabilizes protein hydrophobic bonding because the hydrophobic areas are released from the repulsion of water and the protein tertiary structure is unfolded. 34 At the same time, removal of water destabilizes hydrogen bonding in hydrophilic areas, with the final result of protein denaturation (Fig. 3). This results in inadequate cellular preservation 51, 55, 156 and a possible shift in the intracellular immunoreactivity, as has been reported for some growth factor peptides. 15 There is extensive variability in Ag recognition among tissues fixed in various fixatives and subsequently embedded in paraffin. 5 This different reactivity is intrinsic to the Ag examined and, more specifically, to the epitope recognized by an Ab. There is no “one fixative fits all” situation in immunohistochemistry. Formaldehyde is used mainly because of its reliability in general histology and its low cost; its deleterious effects can usually be countered with AR. 113 In addition, it would be most undesirable for routine diagnostics to have paraffin blocks with tissues fixed in different fixatives over the years. Retrospective studies would be a nightmare. The value of nonformaldehyde fixatives for research purposes has been demonstrated for a variety of Ags. 26, 33, 36, 63, 92, 101, 113, 121, 123, 137, 168, 169
Alcohol fixation. Alcohol will interact with the protein hydrophobic moieties modifying the tertiary structure of the protein (reprinted from Eltoum 34 with permission from the Journal of Histotechnology).
Tissue processing and incubation buffers
Although fixation is very important in determining the outcome of the Ag-Ab reaction, the buffer in which this reaction happens and tissue processing might have some effect in preservation of antigenicity. 31, 50 It has been hypothesized that the combination of cross-linking fixatives with heat and nonpolar solvents used in paraffin embedding modifies the conformation of Ags so that specific epitopes cannot be recognized by Abs that recognize the same epitopes on frozen sections. In other words, in some cases, fixation alone does not prevent immunodetection of the Ag, and tissue processing is the key limiting factor. 31, 40, 44 Decalcification, regardless of the agent used, does not seem to interfere with immunostaining of most Ags, provided the tissues were previously well fixed in formaldehyde. 4 Antibodies are attracted to the epitopes of most glycoproteins and polypeptides—initially through electrostatic charges and subsequently through van der Waals and hydrophobic interactions. 11 The isoelectric point (pI) of polyclonal IgG ranges from 6.0 to 9.5. Monoclonal Abs of IgG class have a similar range of pI values. 50 The Ag–Ab reaction is relatively insensitive to pH in the range of 6.5–8.5 with the use of whole sera as the primary Ab; however, smaller alterations of pH with monoclonal Abs can severely affect binding to Ag. 1, 165 It has been reported that monoclonal Abs perform better in phosphate-buffered saline (PBS) than in Tris buffer, 158 but this result is somewhat puzzling because of the effect of sodium ions (responsible for the ionic strength of a buffer) in PBS that tend to shield negatively charged epitopes, thereby diminishing the attraction of positively charged reactive sites of the Ab, especially with alkaline buffers. 50 Phosphate ions, on the other hand, promote hydrophobic binding, which might explain the suitability of PBS in some instances. Authors of extensive studies conclude that the most suitable diluent buffer for both monoclonal and polyclonal Abs is 0.05–0.1 M Tris buffer (pH 6.0), but there are exceptions. 10, 12, 167 The use of PBS in alkaline phosphatase procedures is not recommended because of its high concentration of inorganic phosphate, which is a competitive inhibitor of this enzyme; Tris buffer is the buffer of choice in this case. 31 Reduction of non-specific background staining as a result of ionic interactions can be achieved by increasing the salt concentration (NaCl) in the buffer from 0.3 to 0.5 M. 111 This increase in ionic strength can also disrupt the binding of specific but low-affinity Abs, so it should be used judiciously.
Ag Retrieval
Fixation modifies the tertiary structure of proteins (Ags), many times making them undetected by specific Abs. This fact is better understood if one remembers that the reaction between the Ag and the Ab depends on the conformation of the former. 50 One of the challenges of IHC is to develop methods that reverse changes produced during fixation. AR producers reverse at least some of these changes. AR is particularly necessary when tissues are fixed in cross-linking fixatives. Approximately 85% of Ags fixed in formalin require some type of AR to optimize the immunoreaction. 115 The need for AR depends not only on the Ag examined but also on the Ab used. Polyclonal Abs are more likely to detect Ags than monoclonal Abs in the absence of AR. 115 The two most common AR procedures used in IHC are enzymatic and heat-based retrieval. A miscellaneous group of AR methods is included.
AR with enzymes
Protease-induced epitope retrieval (PIER) was introduced by Huang. 59, 60 It was the most commonly used AR method before the advent of heat-based methods. Many enzymes have been used for this purpose, including trypsin, proteinase K, pronase, ficin, and pepsin. 8, 62, 90, 102, 107 The PIER mechanism is probably digestion of proteins, but this cleavage is nonspecific and some Ags might be negatively affected by this treatment. 151 The effect of PIER depends on the concentration and type of enzyme, incubation parameters (time, temperature, and pH), and the duration of fixation. 8, 90, 107 The enzyme digestion time is inversely related to the fixation time. 151 It is my and others' preference to optimize a few enzymes rather than use a broad range of enzymes. 7 I use a commercially available, ready-to-use solution of proteinase K that has good activity at room temperature and, therefore, can be used with automatic stainers. The disadvantages of PIER are the rather low number of Ags for which it is the optimal AR method, possible alteration of tissue morphology, and possible destruction of epitopes. 102, 151
Heat-induced epitope retrieval
The heat-induced epitope retrieval (HIER) group of methods has revolutionized the immunohistochemical detection of Ags fixed in cross-linking fixatives (e.g., formaldehyde). HIER was introduced by Shi et al., 132 and it is based on a concept developed by Fraenklen-Conrat and collaborators, 37–39 who documented that the chemical reactions between proteins and formalin can be reversed, at least in part, by high temperature or strong alkaline hydrolysis. The mechanism involved in HIER is unknown, but its final effect is the reversion of conformational changes produced during fixation. Heating can unmask epitopes by hydrolysis of methylene cross-links, 151 but it also acts by other less known mechanisms because it enhances immunostaining of tissue fixed in ethanol, which does not produce cross-links (Fig. 4). 52 Other hypotheses proposed are extraction of diffusible blocking proteins, precipitation of proteins, rehydration of the tissue section allowing better penetration of Ab, and heat mobilization of trace paraffin. 135 Tissue-bound calcium ions might be important in masking some Ags during fixation. Calcium chelating substances (e.g., EDTA) are sometimes more effective than citrate buffer in AR. 95, 106, 160 However, at least on some occasions, calcium-induced changes in the conformation of different proteins might result in increased immunodetection of some Ags, 155 and for many Ags, calcium-induced effect in immunoreactivity cannot be documented. 127, 128 Heating at high temperature (100°C) for a short duration (10 minutes) gives better results than those achieved with a comparatively low temperature for a longer time. 50 However, satisfactory results are obtained in a steamer (90–95°C) with a 20-minute incubation for the majority of Ags needing HIER. A universal AR solution is not available. 50, 61 Thus, several HIER solutions made of different buffers (e.g., citrate, Tris) and with various pH (3–10) have been used. The pH of the solution is important. Some Ags will be retrieved with low pH solutions, others with only high pH solutions; a third group will be retrieved with solutions with a wide pH range. 131 It is others' and my experience that for most Ags, HIER with 0.01 M sodium citrate buffer (pH 6.0) will give satisfactory results and good cell morphology when compared with buffers with higher pH or solutions containing EDTA. 7, 29, 51 Sometimes multiple AR methods are needed to optimize the immunodetection of Ags. 50, 149 In addition, not all Ags benefit from AR, even after prolonged formalin fixation. 135
The degree of fixation can dramatically modify the response of Ags to AR. Unfixed proteins are denatured at temperatures of 70–90°C, whereas such proteins do not exhibit denaturation at the same temperatures when they have been fixed in formaldehyde. 88 In other words, formaldehyde protects from denaturation during AR, which might explain why the immunostaining of partially fixed tissues is sometimes very heterogeneous, despite the supposed even distribution of an Ag throughout the tissue section. 7
The possibility of unexpected immunostaining should always be considered with HIER, particularly with buffers at low pH. 7 When possible, a comparative study of immunoreactivity with fresh frozen and routinely processed paraffin tissue sections is recommended. 127
Miscellaneous AR methods
Pretreatment with concentrated formic acid improves the signal in some IHC tests. 72 Another AR method is incubation of slides in strong alkaline solution, urea, acid solutions, borohydride, and a solution of sucrose. 126, 129
Detection Systems
The Ag-Ab reaction cannot be seen with the light microscope unless it is labeled. Therefore, labels (reporter molecules) are attached to the primary, secondary, or tertiary Abs of a detection system to allow visualization of the immune reaction. A variety of labels have been used, including fluorescent compounds, enzymes, and metals. 80, 146 The most commonly used labels are enzymes (e.g., peroxidase, alkaline phosphatase, glucose oxidase). Enzymes in presence of a specific substrate and a chromogen will produce a colored precipitate at the site of the Ag-Ab reaction. Selection of a detection system is very important, considering that the sensitivity of an immune reaction will depend mostly on the detection system used. Detection systems are classified as direct or indirect methods.
Direct methods
This is the simplest of the immunocytochemical methods. The reaction is a one-step process with a primary Ab conjugated with a reporter molecule (label). 24 Different labels have been used, including fluorochromes, enzymes, colloidal gold, and biotin. 109 The method is quick but lacks sufficient sensitivity for the detection of most Ags in routinely processed tissues (Fig. 5).
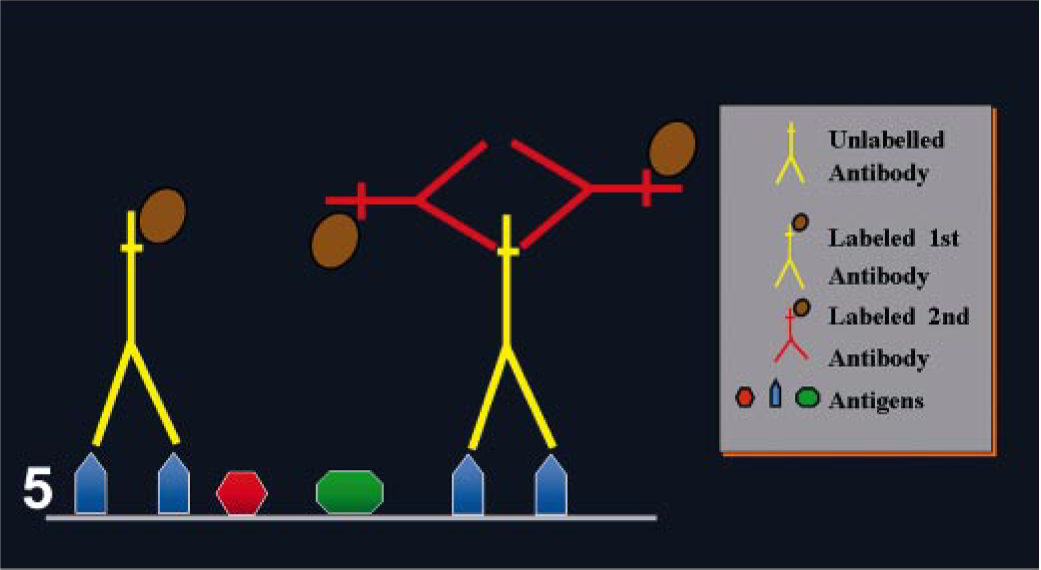
Direct and indirect immunoperoxidase methods.
Indirect methods
The need for more sensitive Ag detection prompted Coons et al. 25 to develop a two-step method. The first layer of Abs is unlabeled, but the second layer, raised against the primary Ab, is labeled (Fig. 5). 109 The sensitivity of this method is higher than a direct method because 1) the primary Ab is not labeled, retaining its activity and resulting in a strong signal and 2) the number of labels (e.g., peroxidase) per molecule of primary Ab is higher, increasing the intensity of reaction. The result is the ability to detect smaller amounts of Ag or to increase the dilution of the primary Ab because at least two labeled Igs can bind each primary Ab molecule. These methods are also more convenient than the direct method because the same secondary Ab can be used to detect different primary Abs, provided the latter are raised in the same species. 109
Avidin–biotin methods
Avidin is a large glycoprotein extracted from egg white that has four binding sites per molecule and high affinity for a low–molecular mass vitamin called biotin. Biotin has one binding site for avidin and can be attached through other sites to an antibody (biotinylated Ab) or any other macromolecule, such as an enzyme, fluorochromes, or other label. 109 The increased sensitivity of avidin–biotin methods results from the larger number of biotin molecules (and therefore label molecules) that can be attached to a primary Ab. 47, 57, 58
One of the most common avidin–biotin methods is the avidin–biotin complex (ABC) method (Fig. 6 and Table 3). In this case, the second Ab is biotinylated and the third reagent is a complex of avidin mixed with biotin linked with appropriate label. The avidin and labeled biotin are allowed to react together for about 30 minutes before being applied, resulting in the formation of a large complex with numerous molecules of label (e.g., enzyme). The proportion of avidin to labeled biotin must be such that some binding sites of avidin to labeled biotin are left free to attach to the biotin on the second Ab. 109 Another commonly used avidin–biotin method is the labeled avidin–biotin (LAB) or labeled streptavidin–biotin (LSAB) (Fig. 7) method, which uses a biotinylated secondary Ab and a third reagent of peroxidase (or alkaline phosphatase)-labeled avidin. The sensitivity of this method is higher than standard ABC. 32
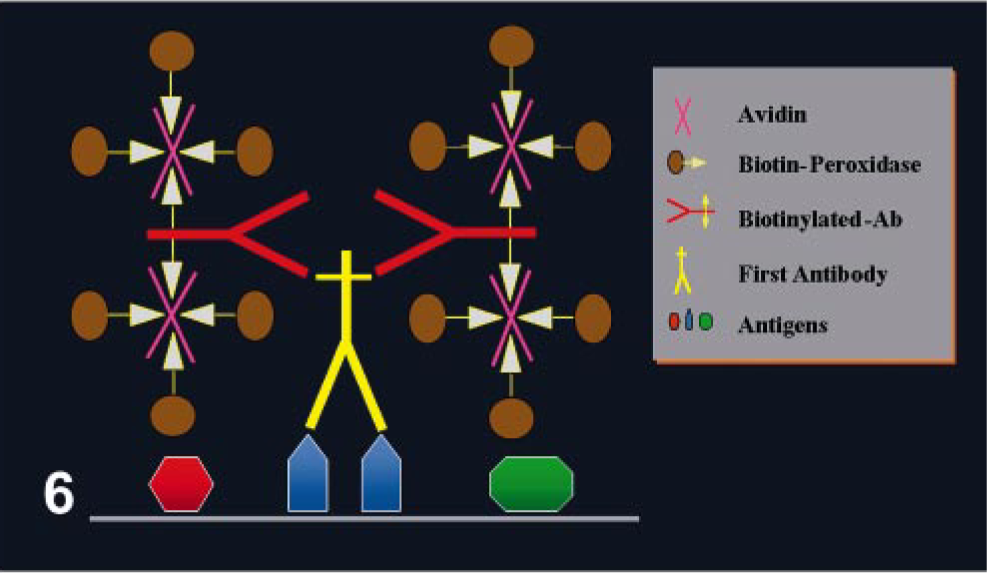
ABC immunohistochemical method.
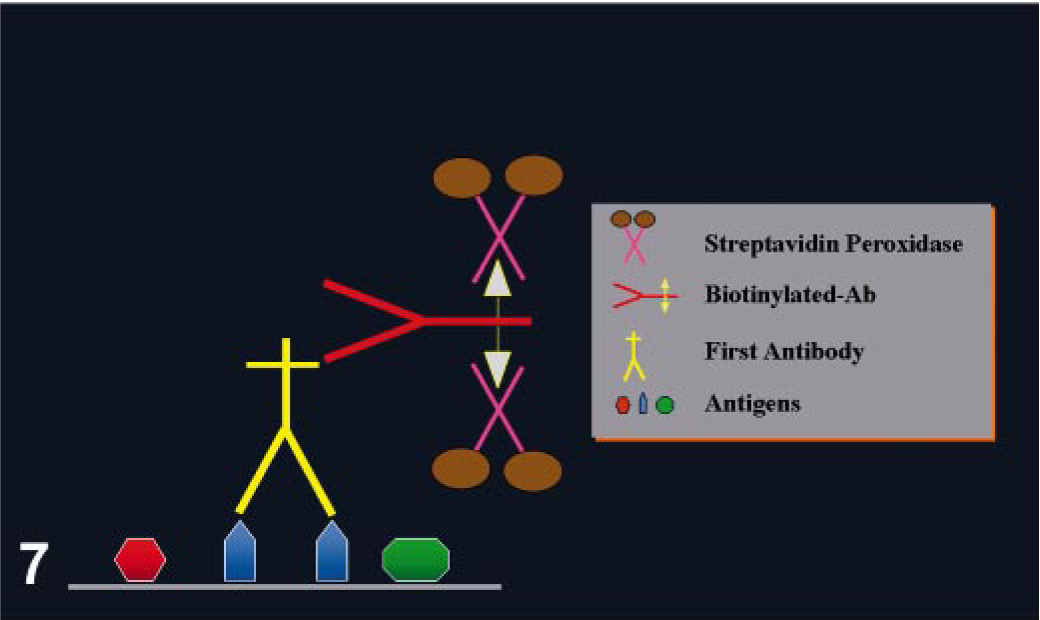
Labeled streptavidin (LSAB)–peroxidase method.
Standard immunohistochemical ABC protocol.∗

∗ The best incubation times for the primary antibody and other reagents should be based on information provided by manufacturers of various reagents and by personal experience. ABC = avidin-biotin complex; HIER = heat-induced epitope retrieval.
† Incubation of the primary antibody can be done at room temperature, 37 C, or 4 C. The best incubation temperature should be determined by trial and error.
One of the main disadvantages of any avidin–biotin system is the possibility of producing high background. Avidin can produce background by binding to lectins in the tissue through its carbohydrate groups and also through electrostatic binding because its pI is 10. This background can be greatly reduced by substituting avidin with streptavidin. Streptavidin, produced by the bacterium Streptomyces avidinii, has a neutral pI, resulting in marked reduction of electrostatic interactions with tissue elements. In addition, because it does not bind lectins, background staining is less likely. However, background from endogenous biotin is still a possibility with streptavidin methods, particularly when harsh AR methods are used. This is particularly common with tissues rich in biotin such as the liver and kidney. 109
Peroxidase–antiperoxidase (PAP) method
This is another indirect method that consists of three layers. 139 PAP has a third layer, which for a rabbit primary Ab is a rabbit antiperoxidase, coupled with peroxidase, in a proportion such that it forms a stable complex (peroxidase–antiperoxidase) composed of two rabbit IgG molecules combined with three peroxidase molecules, one of which they share (Fig. 8). 109 The first and third layers are bound by a second (bridge) layer of Igs (in this example, an anti-rabbit). The key is to add the secondary Ab in excess so as to bind both the primary Ab through one Ab binding site and the PAP complex through the other Ab binding site. This method results in a sensitivity 100–1,000 times higher than in the two-step indirect method. However, PAP is more laborious than the two-step indirect methods. PAP reagents are available to use with goat, mouse, rabbit, rat, and human primary Abs. 146 Although this method was very popular before the advent of avidin–biotin methods, its low sensitivity limits its use today.
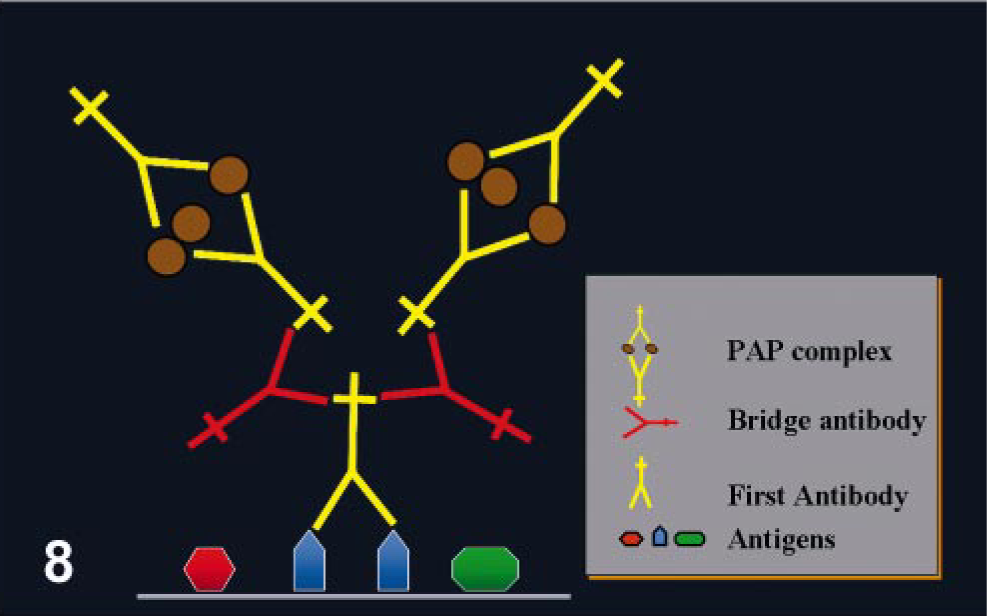
Peroxidase–antiperoxidase (PAP) method.
Polymeric labeling two-step method
This method consists of a compact polymer to which multiple molecules of enzyme and the secondary Ab (specific for the primary Ab) are attached (Fig. 9). The advantages are 1) simplicity compared with the three-step methods, 2) equal or higher sensitivity than ABC or LSAB methods, and 3) lack of background staining because of endogenous biotin or avidin. 105, 122, 130, 146, 153 One disadvantage is that this method is usually more expensive than ABC or LSAB methods. Numerous companies have commercialized polymer-based detection systems (e.g., EnVision™, PowerVision™, Imm-PRESS™).
Two-step polymer-based immunoperoxidase. The secondary reagent has many molecules of label and secondary antigen attached to a polymer backbone.
Tyramine amplification method
This method is based on the ability of tyramine to chemically adhere to a solid substrate (e.g., tissue section) following oxidation/radicalization. 45 Adams 2 adapted this system for immunohistochemistry. The procedure is based on the deposition of biotinylated tyramine at the location of the Ag-Ab reaction, catalyzed by horseradish peroxidase (HRP). Highly reactive intermediates formed during the HRP–tyramine reaction will bind to tyrosine-rich moieties of proteins in the vicinity of the HRP binding sites via the production of free radicals by the oxygen liberated by HRP (Fig. 10). This reaction is short-lived; therefore, the deposition of biotinylated tyramine occurs only in the location at or in immediate proximity to where it is generated. 50 The biotin conjugated to the bound tyramine is subsequently used for the attachment of avidin conjugated to HRP. 50 This method is more complex and laborious because it involves an initial avidin–biotin procedure followed by the tyramine reaction. However, the sensitivity of this reaction can be increased 5- to 10-fold compared with the regular avidin–biotin method; 136 others claim that the increase in sensitivity is even higher. 89 This method is suitable for Ags present at very low amounts in the tissue. However, background can be a problem, particularly with HIER. 48 In this case, more prolonged treatment to quench endogenous peroxidase or endogenous avidin–biotin activities is usually necessary. Modifications of the method with fluoresceinated tyramine result in marked reduction or complete disappearance of background by endogenous biotin. 48, 150
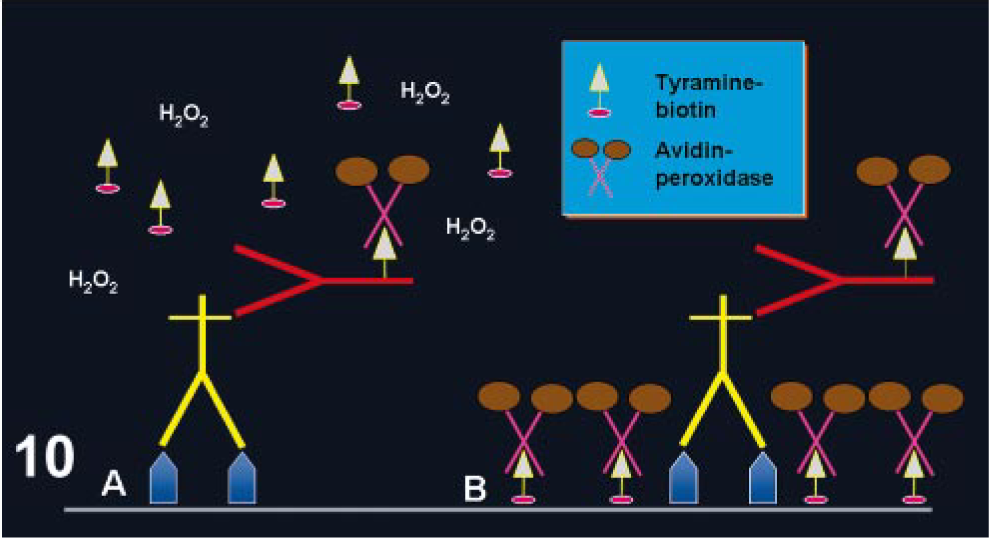
Tyramine amplification method.
Immuno–rolling circle amplification
The aim of this technique is to increase the signal of the immunologic reaction without increasing the noise (background), as can occur with the tyramine amplification method. Rolling circle amplification (RCA) is a surface-anchored DNA replication that can be used to visualize Ags (immunoRCA). This is a combined reaction in which the first part consists of an immunologic reaction (Ag-Ab binding) and the second part is an isothermal nucleic acid amplification with a circularized oligonucleotide probe. 79, 124, 170 This is possible because the primer is coupled to the Ab detecting the Ag. In the presence of circular DNA, DNA polymerase, and nucleotides, the rolling circle reaction results in a DNA molecule consisting of multiple copies of the circle DNA sequence that remain attached to the Ab. The amplified DNA can be detected by hybridization with labeled complementary oligonucleotide probes. 124 The main differences between RCA and polymerase chain reaction is that the former can amplify nucleic acid segments in either linear or geometric kinetics under isothermal conditions, and the product of amplification remains connected to the target molecule. 79, 124 The linear mode of RCA can generate a 105-fold signal amplification during a brief enzymatic reaction. ImmunoRCA has enough sensitivity to detect single Ag-Ab complexes. 124
Other Aspects of Immunohistochemical Reactions
Polyvalent detection systems
Many manufacturers of detection systems offer polyvalent (sometimes called universal) detection systems. The main difference from monospecies detection systems is that the secondary reagent is a cocktail of Abs raised against Igs from different species, allowing one secondary reagent to be used for both polyclonal (e.g., from rabbit and goat) and monoclonal (e.g., from mouse) Abs.
Increasing the sensitivity of Ag detection
The most sophisticated methods described above can detect very small amounts of Ag. However, some of these methods are expensive, and alternatives that use standard methods (e.g., PAP, ABC, LSAB) have been reported, including combining different methods, repeating several steps of the immune reaction, increasing the incubation time of the primary Ab, or enhancing the intensity of the chromogen precipitate. 22, 27, 83, 84, 109, 119, 146
Detection of multiple Ags in a tissue section
Multiple immunolabeling is used to demonstrate the expression of several Ags (cell markers, infectious organisms) in the same or different cells. With the availability of various detection systems (e.g., PAP, ABC, polymer-based methods, commercial dual labeling kits) and different chromogens yielding a variety of colored reactions, multiple immunolabeling is becoming more popular. The key to multiple immunolabeling is that false positive staining resulting from cross-reactivity among different components of the reaction must be avoided. This requires careful planning and the use of multiple controls. Double immunolabeling methods can be divided into sequential and simultaneous staining methods. 148 As a rule, if the primary Abs are made in the same species, sequential dual labeling is necessary; if the primary Abs are made in different species, both primary Abs are added simultaneously.
In sequential staining (same species primary Abs), the second layer of Abs intended for the second Ag could possibly also bind the first Ag (both primary Abs are being produced in the same species) through the primary Ab (Fig. 11). To avoid that, a step previous to the second set of reactions consists of eluting the primary Abs with potassium permanganate or a solution of glycine–HCl for several hours. 31 This treatment might have a deleterious effect on the antigenicity of the second Ag. The elution step (and its deleterious effects on antigenicity) can be avoided by developing the first reaction with a concentrated solution of diaminobenzidine (DAB) that theoretically will block any residual primary Ab of the first immune reaction. 31 Some commercial kits use a blocking step or heat AR step between the first and second sets of immunologic reactions. 148 Sequential double labeling techniques are not recommended in instances in which mixed-color products as a result of colocalization of Ags are expected. 148 In other words, sequential staining is more appropriate for the detection of Ags in two different cell populations or cell locations (e.g., nuclei and cytoplasmic membrane).
Sequential double immunostaining to detect two different antigens with the use of primary antibodies from the same species. The first set of reactions is based on an immunoperoxidase polymeric labeling method. A blocking reagent is added before the immune reaction for the second antigen, which in this case is detected by an immunoalkaline method.
IHC on mouse tissues
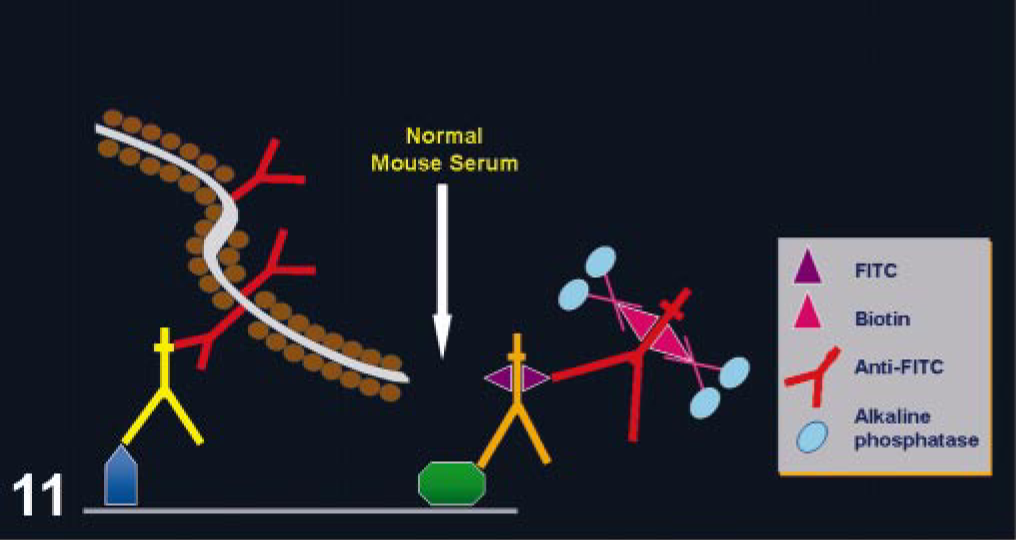
The use of mouse monoclonal Abs on mouse or rat tissues is challenging. Following a standard IHC procedure, background will develop from the binding of the secondary Ab (anti-mouse Igs) to endogenous Igs in murine tissues. 41 This problem has hampered or even precluded the use of IHC with mouse monoclonal Abs in murine tissues. Currently, numerous manufacturers have specific detection systems for mouse tissues, eliminating this problem. One of these methods uses blocking steps before and after addition of the primary Ab; another method preincubates the primary Ab with biotinylated anti-mouse Fab complexes (used as secondary Ab), blocking free binding sites in the complexed secondary Abs with normal mouse serum before adding the Ab mix to the tissue section (Fig. 12). 41, 54, 85
Immunohistochemistry with mouse monoclonal antibodies on mouse tissues. Modifications of the detection system are needed to reduce background. In this case, the primary (mouse antibodies) and secondary biotinylated Fab (anti-mouse antibodies) antibodies are preincubated in vitro. Normal mouse Igs are then added to block free biotinylated anti-mouse Igs. This mixture, containing primary antibodies bound to biotinylated secondary antibodies, is applied to the tissue section (pink) on a glass slide (rectangle labeled 245-04).
Color of the Ag-Ab reaction
The Ag-Ab reaction is not visible under the microscope unless a label is used. The most commonly used labels are enzymes, particularly peroxidase and alkaline phosphatase. Each enzyme has specific substrates and chromogens to produce a colored precipitate. Most common chromogens impart a brown, red, or blue color to the reaction. The choice of enzyme and chromogen will depend on several factors (e.g., intensity of reaction, location of the Ag, presence or absence of endogenous pigments, mounting media used) but often are a matter of personal preference. 151 Many laboratories prefer peroxidase, but alkaline phosphatase (AP) is also a valuable label. Some reports claim that AP methods are more sensitive than similar immunope-roxidase methods. 86, 87 For horseradish peroxidase, 3,3′diaminobenzidine tetrachloride (DAB) is the most commonly used chromogen, giving a brown color, and it is insoluble in organic solvents. When endogenous peroxidase activity is very high (see causes of background below) or melanin pigment is prominent, DAB is not the optimal chromogen, and other chromogens or alkaline phosphatase should be used. 146 3-Amino-9-ethylcarbazole (AEC), another chromogen for peroxidase, produces a red color. Keep in mind that when using this chromogen, coversliping must be made with a water soluble medium (the precipitate will wash out with organic solvents). 4-Chloro-1-naphthol precipitates as a blue product that is soluble in organic solvents. For alkaline phosphatase, 5-bromo-4-chloro-3-indolylphosphate/nitro blue tetrazoliumchloride (BCIP/NBT; blue, permanent media), fast red (red, aqueous mounting media), and new fuchsin (fuchsia, permanent media) are the chromogens most commonly used. Alkaline phosphatase is recommended for immunocytochemistry of cytological specimens. 11
The choice of counterstain will mainly depend on the color of the immune reaction. The counterstain needs to produce enough contrast to avoid confusion with the chromogen precipitate. The most frequently used counterstains are hematoxylin (blue), methyl green (green), and nuclear fast red (red). 151 The counterstain should lightly stain the tissues, without interfering with the chromogen precipitate. In some occasions, particularly with nuclear Ags present in small amounts, counterstain is not recommended.
Causes of Background Staining in Immunhistochemistry
Background is one of the most common problems in immunohistochemistry, and it can seriously affect the interpretation of the immunologic reaction. Here, I list the most common causes of background.
Background produced by hydrophobic interactions of proteins
In aqueous media, hydrophobic interactions between macromolecules occur when their surface tensions are lower than that of water. 11 Hydrophobic forces are key for a successful Ag-Ab binding, but they can also produce unacceptable background. Tissue proteins are rendered more hydrophobic by fixation with aldehyde-containing fixatives as the result of cross-linking of reactive epsilon- and alpha-amino acids, both within and between adjacent tissue proteins. 11 The increased hydrophobicity of proteins during fixation increases the background staining in immunohistochemical procedures; therefore, prolonged fixation in formalin or other aldehyde-based fixatives should be avoided. This background staining from overfixation can be remedied by postfixation with Bouin's, Zenker's, or B5 fixatives. 21 Igs are also very hydrophobic proteins, particularly Abs of the IgG1 and IgG3 subclasses. 11 Aggregation and polymerization leading to increase in hydrophobicity is another problem observed during storage of Igs. Protein–protein interactions between conjugates and polar groups in tissue sections also produce background. 108 Another cause of increased hydrophobicity of Igs is biotinylation of Abs, which can modify their pI. 154
Several methods can reduce hydrophobic binding of Igs and tissue proteins, including diluent buffers with a pH different from the pI of the Ab (particularly for monoclonal Abs); diluents with low ionic strength (low salt concentration); addition of nonionic detergents (e.g., Tween 20, Triton X) or ethylene glycol to the diluent; or raising the pH of the diluent used for polyclonal Abs. 11, 69, 108 However, probably the most common method to reduce background from hydrophobic interactions is the use of blocking proteins prior to incubation of the primary Ab (Fig. 13). Classically it has been performed with Igs of the same species to the secondary link or labeled Ab; however, bovine serum albumin, fish gelatin, fetal calf serum, nonfat dry milk, and, more recently, casein can be used. 28, 75, 144 Casein appears to be more effective than normal serum to block hydrophobic background staining. 144
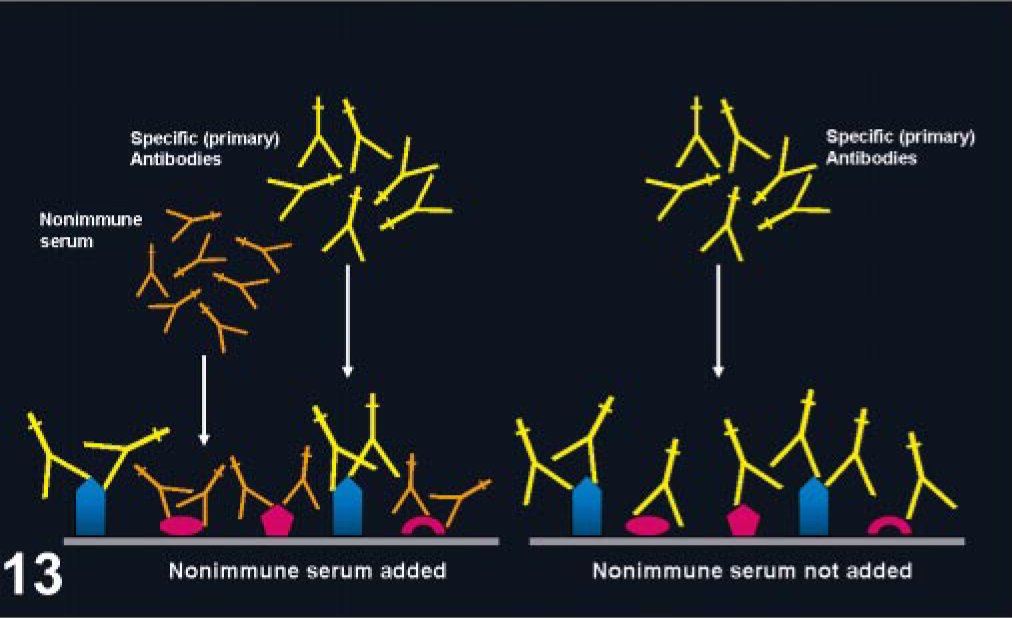
Blocking nonspecific background with normal serum. Normal serum added before the primary antibody (left) will block nonspecific binding of the primary antiserum. When blocking is not used (right), the primary antibody can bind unrelated antigens (the blue bullets are the antigens targeted by the primary antibody) in the tissue section producing background.
Background produced by ionic and electrostatic interactions
Ionic interactions are one of the main forces that control Ag-Ab interactions, but they also contribute to nonspecific background. The pI of the majority of Abs ranges from 5.8 to 8.5. 11 At the pH commonly used in diluent buffers, Abs can have either net negative or positive surface charges, and ionic interactions between Igs and tissue proteins can be expected if the latter possess opposite net surface charges. Nonimmune binding of Igs to tissues or cells with negative charge (e.g., endothelium, collagen) can be blocked effectively by diluent buffers with high ionic strength. 11 AR with 1% zinc sulfate, 0.01 M citrate (pH 6.0), or 0.01 M Tris (pH 9.0) can result in nonspecific nuclear staining. The cause of this nonspecific binding is unclear, but it has been hypothesized that a combination of electrostatic and polar (electron acceptor/electron donor) forces are involved in this problem. 11, 159 Detection of some Ags is improved if the ionic strength of the fixative solution is increased. 17 Solving nonspecific background staining in immuno-histochemistry becomes more complicated with the realization that, many times, this nonspecific staining is the result of a combination of ionic and hydrophobic interactions, and, as previously mentioned, remedies for one type of interaction can aggravate the other. 11
Endogenous peroxidase activity
Enzyme activity naturally present in red blood cells (pseudoperoxidase), granulocytes (myeloperoxidase), and neurons can react with DAB to produce a brown product indistinguishable from specific immunostaining. 31 Although endogenous peroxidase activity is destroyed almost completely during formalin fixation, pretreatment of tissue sections with a diluted solution (0.003–3%) of H2O2 in methanol will further reduce or completely abolish pseudoperoxidase activity of red blood cells and peroxidase activity in myeloid cells. 140, 142 In tissue sections with abundant hemorrhages or with acid hematin, a stronger (10%) solution of H2O2 might be needed to remove this endogenous activity, 3, 141 or a longer incubation in less concentrated solutions. 31 Use of H2O2–methanol is not recommended for specimens in which cell surface Ags are to be detected; also, the use of methanol might detach frozen sections from the glass slide. 11 In these instances, endogenous peroxidase activity can be suppressed by a mixture of H2O2 (0.3%) in 0.1% sodium azide 77 or H2O2 diluted in distilled water. Endogenous peroxidase background might be more apparent with the use of very sensitive detection systems. To reduce this background, the H2O2 concentration needs to be increased or an alternative enzyme needs to be used. Other methods that have different effects on Ag detection also inhibit endogenous peroxidase activities. 31
Endogenous alkaline phosphatase
The use of AP as a reporter molecule is increasing because it facilitates double immunolabeling and avoids most of the problems of endogenous peroxidase activity found in hematopoietic tissue. Two isoenzymes of AP in mammalian tissues can produce background staining with AP methods: intestinal and non-intestinal forms. The nonintestinal form is easily inhibited by 1 mM levamisol (
Avidin and biotin as sources of background
The high ionic attraction of basic egg white avidin for oppositely charged cellular molecules such as nucleic acids, phospholipids, and the glycosaminoglycans in the cytoplasm of mast cells could result in nonspecific binding. 104, 162 Egg white avidin has an pI at pH 10.0 and has a basic positive charge at the almost neutral pH used in immunostaining. This non-immune binding can be prevented by preparing ABC or LAB solutions at pH 9.4 instead of at 7.6 or by adding a 5% solution of nonfat dry milk. 18, 28 Substituting avidin from egg white with streptavidin (from Streptomyces avidinii), which has a pI at a pH of 5.5–6.5, reduces significantly the nonspecific binding in IHC methods. 13, 18, 96, 162, 164
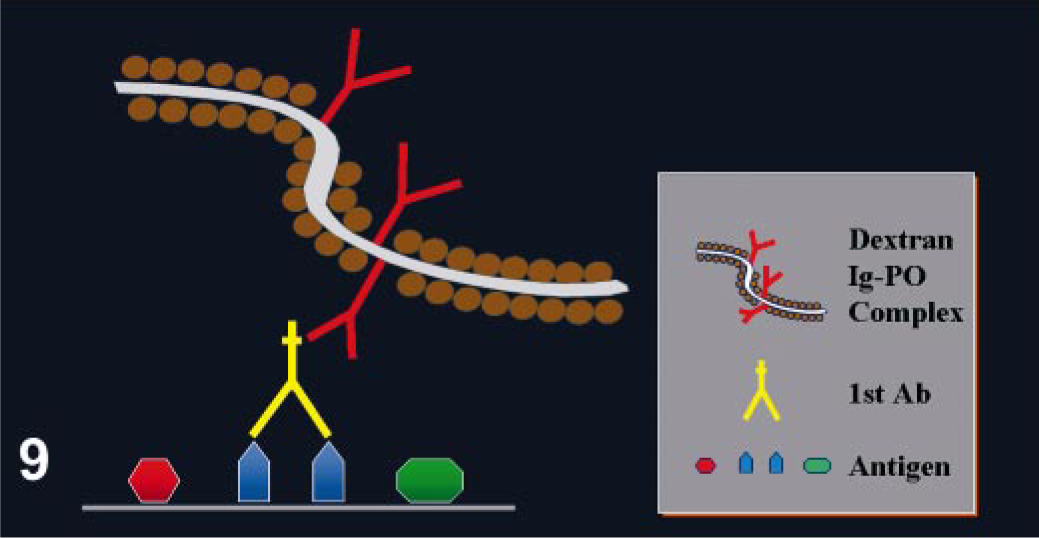
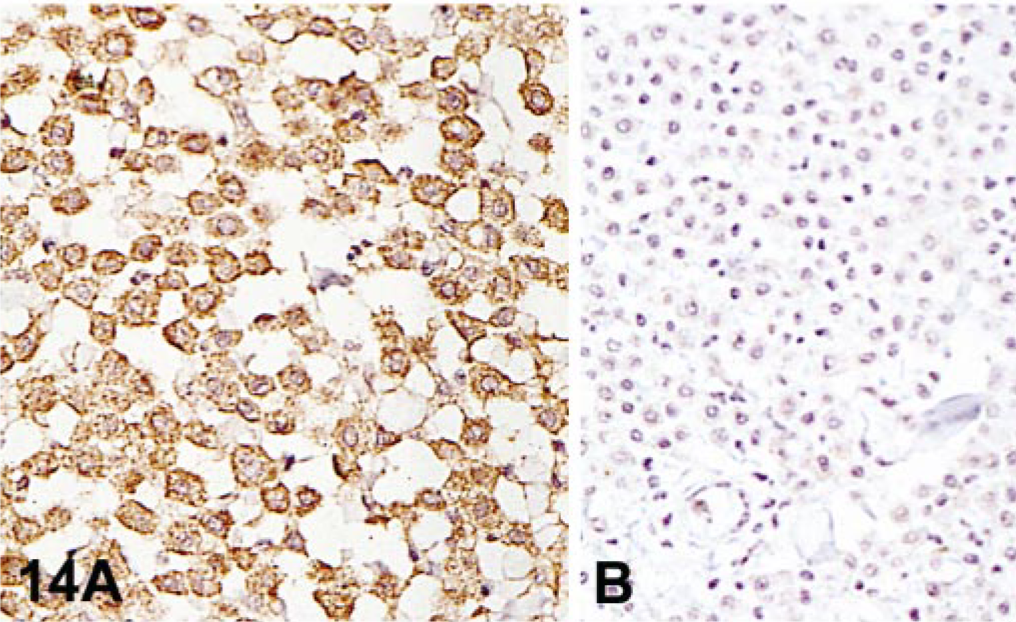
Endogenous biotin is widely dispersed in mammalian tissues, particularly in liver, lung, spleen, adipose tissue, mammary gland, kidney, and brain. 13, 19, 96, 162, 164 Background from endogenous biotin is greatly diminished after formalin fixation but can be pronounced in frozen sections. 11 Some tumor cells can have intranuclear inclusions containing biotin; endometrial cells during gestation and postpartum can have also intranuclear biotin inclusions. 133, 163, 166 Harsh heat-based AR methods expose endogenous biotin in formalin-fixed tissues (Fig. 14). 19 Binding of avidin used in detection systems to endogenous biotin can produce strong background and needs to be inhibited. 31 This binding can be suppressed with alkaline buffers, preincubation of tissue sections with unlabeled avidin and biotin (Figs. 15, 16), or incubation with nonfat dry milk. 6, 28, 66, 161 Some commercial kits containing 0.1% of streptavidin and 0.01% of biotin block this endogenous activity. This can interfere with interpretation of nuclear Ag staining (proliferation markers, herpes infections).
Background from endogenous avidin–biotin activity.
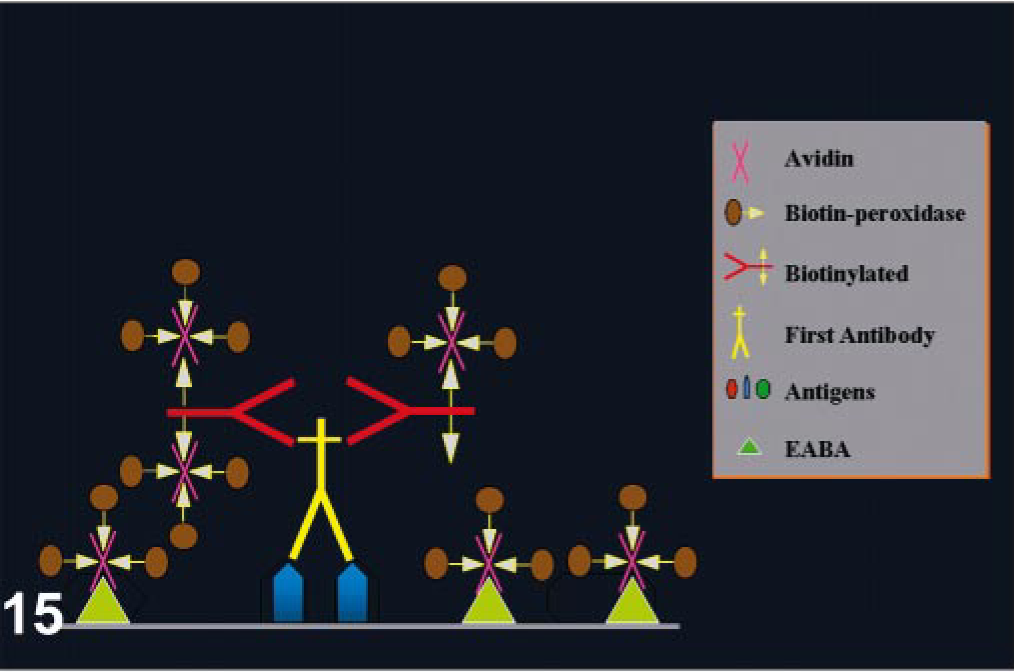
Endogenous avidin–biotin activity (EABA). In this case, avidin–biotin–peroxodase complexes will nonspecifically bind tissues, producing strong background.
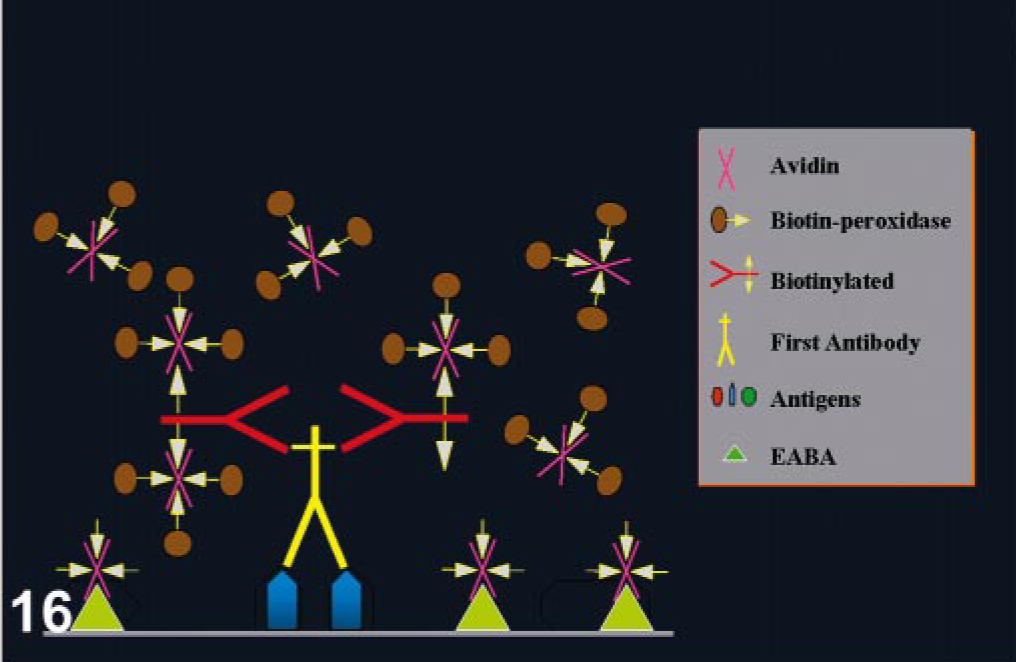
Blocking EABA. The addition of unlabeled avidin and biotin before the immune reaction will block nonspecific background by EABA.
Free aldehydes
False positive staining might result from the non-specific attachment of conjugated Abs to free aldehyde groups introduced by aldehyde-containing fixatives present in the tissue. This problem is more common when glutaraldehyde is being used, but prolonged fixation in formaldehyde can also produce free aldehydes. Different compounds abolish this binding (sodium borohydride, ammonium chloride, ammonium carbonate buffer, lysine, glycine). 31 Keep in mind that some of these treatments might modify antigenicity. 30, 31
Fc receptors
Fc receptors of mononuclear blood cells can bind to IgG of antisera, but this is not usually a problem with routinely fixed paraffin-embedded tissue because Fc receptors are destroyed during this process. 31 However, nonspecific adherence of the primary antiserum to tissues could occur with lightly fixed, frozen sections of lymphoid tissue or cytological preparations containing cells with Fc receptors. Nonspecific staining can also happen in paraffin sections because of attraction of the Fc portion of Igs to basic groups present in collagen fibers. 157 The use of F(ab′)2 fragments of the Igs instead of the whole Ig molecule eliminates nonspecific background from Fc receptors or the Fc portion of Igs. 16
Nonspecific Ag diffusion (sequestration)
Diffusion of soluble proteins from their constituent cells and their nonspecific sequestration by other cells of different lineage or the cell interstitium is a common problem in thyroglobulin detection; it can be observed also with myoglobin, glial fibrillary acidic protein, and other cellular proteins (Fig. 17). 35, 71, 138 Interstitial staining is frequently seen in tissues with a high concentration of Igs in blood plasma that perfuses to the tissue prior to fixation. 11 AR has improved our ability to detect many Ags. However, sometimes unexpected reactions are observed (Fig. 18).
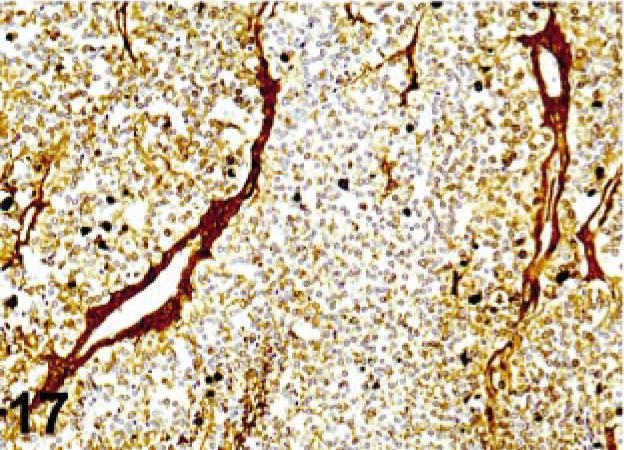
Antigen diffusion background; thyroid carcinoma. The strong staining of connective tissue and interstitium in this section stained with thyroglobulin antibody is the result of diffusion of thyroglobulin from inadequate fixation.
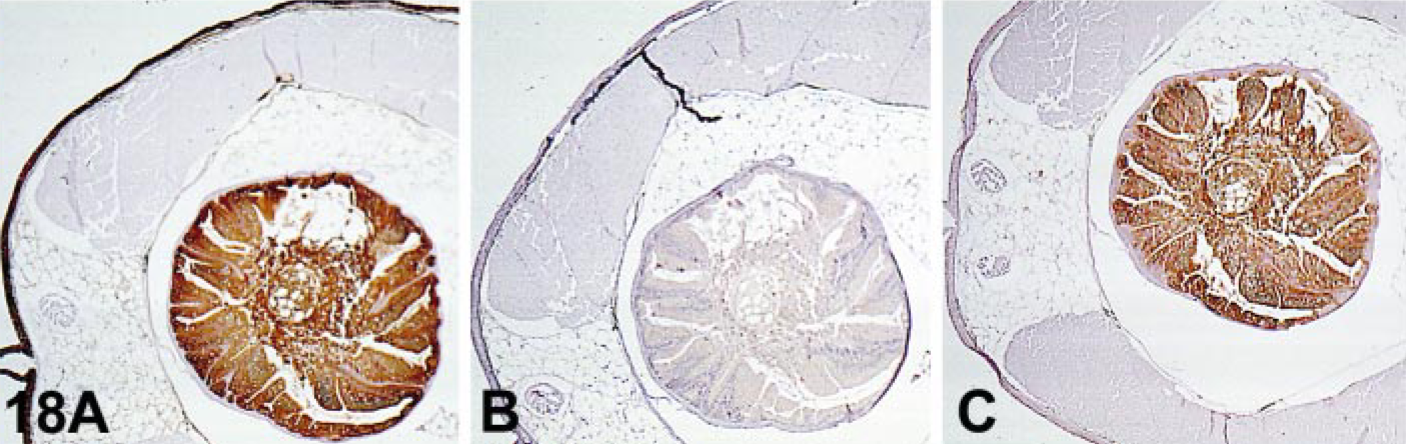
Effect of AR on immunostaining.
Pigments
Tissues with abundant melanin or ferrous pigment, such as hemosiderin, can reduce the signal-to-noise ratio of immunocytochemical reactions. 31 If the pigment is present in the same cell as the Ag examined, interpretation might be impossible. Alternatively, a detection system producing a different colored precipitate can be used. Another possibility is the use of potassium permanganate to block melanin, but this can damage certain epitopes. 31 An alternative to this treatment is the use of Giemsa stain or Azure B dye as a counterstain after the immunoreaction is done; melanin will stain green or blue-green, and DAB product will remain brown. 70, 116 For hemosiderin, a 1% solution of dithionite in pH 5.0 acetate buffer for 5 minutes completely eliminates hemosiderin and also lowers background. 31
Footnotes
Acknowledgements
I appreciate the revision of this manuscript by Dr. HogenEsch and Dr. Miller, Department of Veterinary Pathobiology, Purdue University.
1Present address: Animal Disease Diagnostic Laboratory, Purdue University, West Lafayette, IN.