
Abstract
Seventy-four 9-week old female C57BL/6J mice housed in a conventional facility were manipulated to induce experimental autoimmune encephalomyelitis, among which 26 developed clinical signs including lethargy, absence of defecation, and abdominal distension. By gross necropsy examination, there was distension of the cecum and colon with fecal impaction. By histologic examination, there was severe ulcerative and proliferative typhlocolitis. Fecal ELISA confirmed the presence of toxins A and B of Clostridium difficile. Alteration in immune status of the immunocompetent mice, due to stress caused by experimental manipulation or autoimmune disease, may have led to intestinal dysbiosis, followed by opportunistic infections resulting in C. difficile–associated disease. This report brings to light the occurrence of the disease in immunocompetent laboratory mice during experimental manipulations associated with alteration in immune status, and it discusses potential hazards associated with conventional housing within a hospital-associated research institute.
Keywords
Seventy-four 9-week-old female C57BL/6J mice, housed in open top cages in a conventional facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, were manipulated to induce experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. The induction protocol included administration of 500 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) intraperitoneally on days 0 and 2 and emulsions of 400 μg of MOG 35-55 peptide (myelin oligodendrocyte glycoprotein, Penn State College of Medicine Core Facility) with Freund’s complete adjuvant (Sigma-Aldrich, St. Louis, MO), supplemented with 5 mg/mL of heat-inactivated Mycobacterium tuberculosis (Difco, Kansas City, MO), subcutaneously on days 0 and 7. Intraperitoneal injections of vehicle (phosphate buffered saline), naltrexone (0.1 mg/kg), or [Met 5 ]-enkephalin (10 mg/kg) were administered on a daily basis. Beginning on day 2, mice were observed for behavioral signs of EAE, including loss of tail tonus, gait abnormality, and/or hind limb weakness. Because the study protocol required frequent specialized behavioral testing of the mice within the investigator’s laboratory, mice could not be housed in a barrier-type facility. The mice exhibited clinical signs, including lethargy, hunched posture, shallow breathing, absence of defecation, and abdominal distension, culminating in death or euthanasia beginning at day 8 from the initiation of the induction protocol. Morbidity and mortality were observed in a total of 26 mice from all treatment groups over a 3-week period. Distended loops of bowel were palpable in the caudal abdomen of both live and dead mice. Six moribund mice (mice Nos. 1–6), 2 of which demonstrated behavioral signs of EAE, were euthanized, and at gross necropsy, there was marked distension of the cecum and colon and, to a lesser extent, the distal small intestine, by a large quantity of relatively dry fecal material (Fig. 1). The mesenteric blood vessels were acutely congested. The stomach and proximal small intestine were empty with no gross lesions. There was no other gross pathology noted. Similar gross gastrointestinal lesions were observed on cursory gross examination of the other 20 mice, 12 of which were necropsied by research personnel before the case came to the attention of the clinical medicine staff.
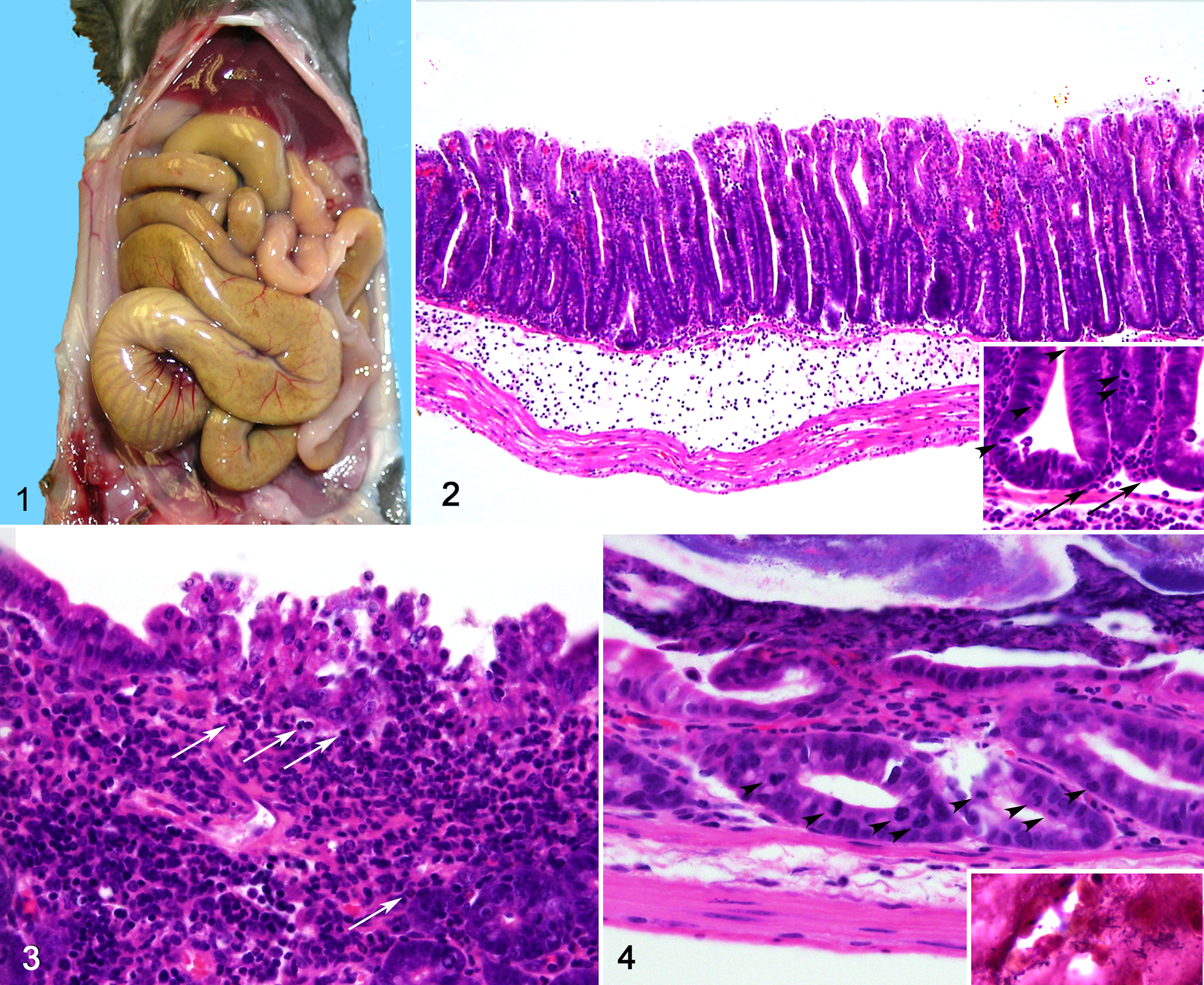
The lumen of the cecum and colon is markedly distended and impacted with a large quantity of firm fecal material, with dilation of the distal small intestine. Scale in centimeters.
To exclude well-characterized mouse pathogens as possible etiologic agents, diagnostic tests were performed on only the affected mice. Aerobic culture of the cecal contents was positive for Pseudomonas aeruginosa in 3 of 4 mice (Nos. 1, 3, and 4) and negative for Citrobacter rodentium and Salmonella spp. in 4 of 4 mice (Nos. 1–4). Serology results were negative for all tested pathogens, including Clostridium piliforme, mouse hepatitis virus, and mouse norovirus (RADIL mouse comprehensive plus panel, mouse 1). Direct fecal examination tested negative for pinworms on 4 of 4 mice (Nos. 1–4). One mouse tested positive by fecal polymerase chain reaction for Helicobacter (No. 5, Helicobacter hepaticus only); 2 other mice tested negative (Nos. 1 and 6). A pooled fecal sample from 3 mice (Nos. 2–4) was tested for Clostridium difficile toxin using a rapid, qualitative, horizontal flow enzyme immunoassay kit (Immunocard Toxins A and B, Meridian Biosciences Inc., Cincinnati, OH), following manufacturer’s recommendation. Briefly, the sample was mixed with sample diluent and enzyme conjugate and then dispensed in the Immunocard ports. Card ports were washed with the wash buffer, followed with substrate dispensation. Results were read after a 5-minute incubation at the room temperature. The sample tested positive for C. difficile toxins A and B.
For histopathology (mice Nos. 1–6), tissues were fixed in 10% neutral buffered formalin. Tissues were processed in an automated Tissue-Tek VIP processor and paraffin-embedded with a Tissue-Tek TEC embedding station. Sections were cut at 6 μm for routine hematoxylin and eosin staining. Histologically, lesions were limited to the cecum and colon with sparing of the small intestine. Crypts were markedly elongated, crowded, and hyperplastic with frequent mitoses, cytoplasmic basophilia, high nuclear:cytoplasmic ratio, and nuclear hyperchromasia. The submucosa was markedly edematous with large numbers of nondegenerate neutrophils and fewer macrophages in a pale eosinophilic homogeneous material (Fig. 2). Multifocally small clusters of surface epithelial cells were degenerate or necrotic and exfoliating (erupting) in tufts into the lumen (Fig. 3). There were frequent erosions or ulcerations of the mucosal epithelium with infiltration of large numbers of degenerate neutrophils and fewer macrophages in the mucosa and submucosa. In some foci, the underlying stroma was expanded by plump and closely spaced fibroblasts in an immature matrix (fibroplasia). Occasionally throughout the sections, there was an adherent pseudomembrane composed of fibrillar beaded protein (fibrin), degenerate neutrophils, and necrotic cellular debris adhered to the colonic mucosal surface (Fig. 4). There were low numbers of inflammatory cells in the perivascular spaces of the muscularis and serosa/mesentery. No bacteria morphologically consistent with C. piliforme were identified on Steiner-Chapman or Wright-Giemsa stains. No protozoal or fungal agents were identified on periodic acid–Schiff stain.
The clinical presentation and gross and histologic lesions in the cecum and colon, combined with supplemental diagnostic testing results, support a diagnosis of proliferative and ulcerative typhlocolitis caused by C. difficile. Lesions are consistent with those reported previously in mice with natural and experimental infections. 3,8 C. difficile is a spore forming Gram-positive anaerobic bacillus. It is identified as the cause of C. difficile–associated disease (CDAD), characterized by pseudomembranous colitis and diarrhea. This is an opportunistic nosocomial disease commonly resulting from use of antibiotics that cause disruption of normal intestinal flora. In healthy individuals, the competition for nutrients with normal intestinal flora suppresses the growth of C. difficile. 16 A healthy immunocompetent mouse may harbor low numbers of toxigenic strains of C. difficile in the cecum and colon. 11 Soon after dysbiosis, C. difficile organisms rapidly multiply within the lumen and colonize the intestine with production of toxins. The toxins bind to the apical side of the mucosal epithelial cells, become internalized, and act on guanosine triphosphate–binding rho proteins to cause disruption of actin cytoskeleton and tight junctions, resulting in destruction of intestinal epithelium, cell death, and production of inflammatory mediators that could result in septic shock and death. In laboratory animals, experimental antibiotic-associated colitis caused by C. difficile has been described in hamsters, 1 guinea pigs, 10 and rabbits. 6 Natural infections have been reported in hamsters, piglets, and foals. 14 This is a rare spontaneous disease in mice, with a single case report in a colony of immunocompromised C3SnSmn.CB17-Prkdcscid /J mice. 8 However, infections in mice may be underreported due to the absence of C. difficile monitoring from routine surveillance programs, as well as the difficulty in establishing a diagnosis of CDAD.
Although diarrhea is a common clinical sign of C. difficile infection, this may not be manifested in conditions such as ileus, as noted in this cohort of mice. Minimal or no diarrhea has been observed in a small percentage of C. difficile–infected patients with ileus. 4,7 Furthermore, reduced production of feces was noted in the previously reported spontaneous C. difficile outbreak in mice. 8
Studies of experimental infections in mice have demonstrated that the use of antibiotics that suppress the normal intestinal flora, as well as immunosuppressive drugs such as cyclosporine, promote C. difficile infection. 7 A number of established mouse models of C. difficile infection have been developed using a mixture of antibiotics 3 and cyclosporine 7 in C57BL/6 and BALB/c mice, respectively. Other models include hamsters and gnotobiotic mice. Diagnostic tests include detection of toxin A (enterotoxin) and toxin B (cytotoxin) in feces using ELISA or a fecal polymerase chain reaction assay for detection of plasmids encoding the toxins. Cell cytotoxicity assay and culture of organisms from fecal samples are alternate diagnostic tests. 4
It is unclear what caused the nominally immunocompetent mice in this study to develop CDAD. Immune modulation and stress resulting from the experimental induction of EAE may have altered intestinal flora, leading to dysbiosis with establishment of C. difficile infection, including toxin production and secondary colonization by opportunistic bacteria such as P. aeruginosa, a common organism present in non-acidified drinking water. Although P. aeruginosa was isolated from the fecal culture, it is not known to be associated with gastrointestinal disease in mice. Previous studies in mice have demonstrated that the release of norepinephrine, an effector of stress response, can cause alteration of normal gut flora, especially in the cecum, that possibly increases the risk for infectious diseases. 12 Alteration in the gut immune status, including innate and adaptive immunity, could bring about changes in the gut microbial environment. Such an alteration in gut immunity will alter the fine balance of host-commensal homeostasis that results in growth of opportunistic pathogens. Another study demonstrated that the experimental transmission of C. difficile to Myd88tm1Aki mice deficient in innate immune response resulted in severe intestinal disease, while immunocompetent control mice developed a self-limiting disease, thus demonstrating the role of innate immunity in C. difficile infection. 11 Studies have shown an inverse correlation between secretory IgA, a component of adaptive immunity, and release of norepinephrine. 5 In times of stress, a decreased secretory IgA will not effectively prevent the growth of opportunistic pathogens. The EAE induction regimen in this mouse cohort could have severely altered the host immune response.
Previous studies in human patients have established a positive correlation between C. difficile infection and incidence of autoimmune hepatitis independent of the use of immunosuppressive drugs. Risk factors such as autoimmune disorders may impair protective immunity, thus increasing susceptibility to opportunistic agents such as C. difficile. 15 A similar scenario with immune dysregulation and impaired protective immunity could have occurred in the EAE-induced mice, increasing susceptibility to C. difficile. Triggering abnormal immune responses to components of the normal intestinal microflora may contribute to the development of intestinal dysbiosis. Besides EAE induction regimen, the colonization by Helicobacter is a possible confounding factor in disrupting the cecal microflora. This change in diversity of microbiota may result in alteration of local immune response, thus rendering the mice susceptible for C. difficile–associated diarrhea. 2,9 Note, however, that despite a relative high incidence of murine Helicobacter spp. infections, particularly in academic settings, CDAD has not heretofore been recognized as a sequela.
C. difficile is a ubiquitous sporulating organism that is highly resistant to several disinfecting agents and has a documented ability for facile widespread dissemination. 11 Toxigenic C. difficile organisms can be present endogenously within the gut in a carrier state, or transmission can occur from external sources such as hospitals. Infections are more prevalent in hospital settings, with a high probability of detecting toxigenic strains from the hospital floors, equipment, and hands of asymptomatic personnel that come in contact with patients suffering from CDAD. 4 Housing mice in open top cages within a conventional facility that is closely associated with hospital increases the risk of infections to mice with potentially altered immune status due to experimental manipulation. A low inoculating dose of spores can result in colonization and gastrointestinal symptoms of variable severity. Although an experimental inoculation of an human-adapted C. difficile strain, M68, has been demonstrated to cause clinical signs in mice, 11 in this report it was not determined if the C. difficile was of human or animal origin. Strict barrier housing with proper protective equipment for personnel and sanitary procedures using sporicidal agents should effectively prevent any contamination. Routine monitoring of barrier room sentinel mice for C. difficile toxigenic strains can ensure that the room is C. difficile negative, and ensuring that the mice being received from the commercial source are free of C. difficile toxigenic strain will certainly help.
In conclusion, this study underscores the risk factors associated with experimental manipulation in laboratory animals that potentially make apparently immunocompetent mice susceptible to C. difficile infection. Having previously demonstrated the role of intestinal commensal microflora in the development of EAE disease, 13 this study brings to light the alteration in the intestinal flora due to EAE induction, which potentially has an impact on the severity of the disease manifestation.
Footnotes
Acknowledgements
We wish to thank Weifang Lin and Ellen Mullady for histology preparation and Sherry Cooper and Nancy Harner for microbiology and clinical pathology support.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This research was supported in part by funding from The Paul K. and Anna E. Shockey Family.