
Abstract
The p53 tumor suppressor gene (TP53) is the most frequently altered gene in human cancer. Mutation of the gene has been shown to be an important mechanism of p53 pathway inactivation in a variety of human brain tumors, particularly those of astrocytic origin. Genomic DNA from a series of 37 glial and 51 nonglial canine brain tumors was sequenced to determine the frequency of TP53 gene mutations involving exons 3–9. Exonic mutations were found in 3 of 88 tumors (3.4%) and specifically in 1 of 18 astrocytic tumors (5.5%). This is markedly lower than that reported in comparable human tumors, suggesting that alternative mechanisms of p53 inactivation are likely to be present if p53 function contributes significantly to oncogenesis in canine brain tumors.
The TP53 tumor suppressor gene is constitutively expressed in almost all cell types, and it acts as a transcription factor involved in a multitude of functions, including cell cycle control, programmed cell death, senescence, differentiation and development, DNA repair, and maintenance of genomic stability (reviewed in Hainaut and Hollstein 11 ) As such, it is critically involved in oncogenesis, is the most frequently altered gene in human cancer, and has been implicated in the generation and progression of several types of brain tumors. *
The TP53 gene contains 11 exons (the first exon is noncoding), and it has a high degree of sequence similarity across mammalian species. The canine p53 product (381 amino acids) is 81% identical to human p53 (393 amino acids), with the difference primarily due to an extra 13 amino acids in human exon 4. 7
The majority of human TP53 tumor mutations are single base pair missense mutations, with over 90% of identified mutations occurring in the p53 DNA-binding domain (exons 4–8). 11 In most cancers, one allele is lost, while the other carries a mutation; however, loss or inactivation of the wild-type allele is not necessary in all cases. Some mutated p53 proteins have been shown to inactivate wild-type p53 in a dominant negative manner, 12 , 25 and some mutants have gain of function characteristics resulting in pro-oncogenic effects. 38
As with human cancer, TP53 mutations have been reported in a variety of canine cancers, including mammary carcinoma, 7,42 osteosarcoma, 24,34 lymphoma, 34,39 thyroid carcinoma, 9 papilloma, 23 and various soft tissue sarcomas. 27,34 Reports of TP53 mutations in canine brain tumors are limited to a single mutation in a series of 12 astrocytic tumors. 36
Mutation and/or allelic loss of the TP53 gene has been described in up to 60% to 80% of low-grade (II) and anaplastic (III) astrocytomas in humans, where it is thought to represent an early oncogenic event. 33,35,40,43,44 Occurrence of TP53 mutations in high-grade (IV) astrocytomas/glioblastoma multiforme (GBM) depends on whether the tumor is a primary (de novo) GBM, where the incidence is low (∼10%), or a secondary GBM that has developed from a lower-grade tumor (∼ 65%). 33,35,41,43,44 TP53 mutations in other human primary brain tumors are relatively uncommon based on published data and the IARC TP53 Database (version R15, November 2010) from the International Agency for Research on Cancer: oligodendroglioma (up to 18%), meningioma (up to 13%), choroid plexus tumors (up to 18%), and ependymoma (up to 4%). †
The purpose of this study was to further define the potential role of the TP53 gene in canine primary brain tumors by defining the frequency of TP53 gene mutations in a series of canine glial and non glial brain tumors.
Materials and Methods
Sample Collection
All tumor tissue was obtained from surgical biopsy/resection specimens or at necropsy from clinical cases presented to the Veterinary Medical Teaching Hospital, University of California, Davis, or the Veterinary Teaching Hospital, University of North Carolina. Samples of tumor from necropsy were collected within 20 minutes of death and snap-frozen in liquid nitrogen. Surgical samples were similarly stored following collection. Samples of adjacent tumor tissue were processed for routine paraffin embedding and histological analysis whenever fresh tissue was collected in liquid nitrogen. Nontumor genomic DNA (gDNA) was collected from animals with tumors, when available, by sampling whole blood, liver, or temporalis muscle. gDNA was sequenced from 7 cell lines: 3 previously characterized canine mammary carcinoma cell lines, which were used as positive controls 37 (CMT3, CMT7, CMT8), and 4 canine glioma cell lines (J3T, 2 J3T-Bg, SDT-3, G06A). The J3T-Bg glioma cell line was derived in our laboratory from the canine J3T cell line following passage as a subcutaneous tumor through Biege-Nude-xid mice (Taconic Farms Inc., Hudson, NY). The SDT-3 and G06A cell lines were derived in our laboratory from spontaneous canine astrocytomas (grade IV). All tumors were classified by a board certified pathologist (R.J.H.) essentially according to the international World Health Organization classification of human tumors of the central nervous system. 19 Grading was not possible in some small samples obtained from stereotactic computed tomography-guided biopsy procedures.
gDNA Extraction
gDNA was extracted from tumor and control tissues using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, California). DNA integrity, concentration, and purity for all samples were assessed by agarose gel electrophoresis and spectrophotometry.
TP53 Polymerase Chain Reaction
Exons 3–9 of the canine TP53 gene were amplified using 4 sets of primers: flanking exons 3 and 4, 5 and 6, 7 and 8, and 9, respectively. At least 1 primer for each product was intronic (Table 1). All primers were designed using Primer3 software (Biomatters Ltd, Auckland, New Zealand) and were manufactured by Eurofins MWG Operon (Huntsville, Alabama). Amplification was carried out in 10 μL reactions containing 1× polymerase chain reaction (PCR) buffer, 1.00mM MgCl2, 0.2mM of each of 4 dNTPs, 1μM each of sense and antisense primers, 0.0225 U/μl of Thermoprime Plus DNA Polymerase, and 100 ng of gDNA (all reagents from Thermo Fisher Scientific, Rockford, Illinois). Reactions were performed using a Mastercycler Gradient (Eppendorf, Hamburg, Germany) set to 3 minutes at 94°C followed by 34 cycles of 1 minute at 94°C, 1 minute at 60°C (exons 3, 4, 7, 8, 9) or 62°C (exons 5, 6) and 1 minute at 72°C with a final elongation period of 10 minute at 72°C. Quality of PCR amplicons was analyzed using agarose gel electrophoresis, and quantity was estimated by comparison to a 1kb+ ladder (Invitrogen, Carlsbad, California).
gDNA Polymerase Chain Reaction Primers for Canine TP53 Exons
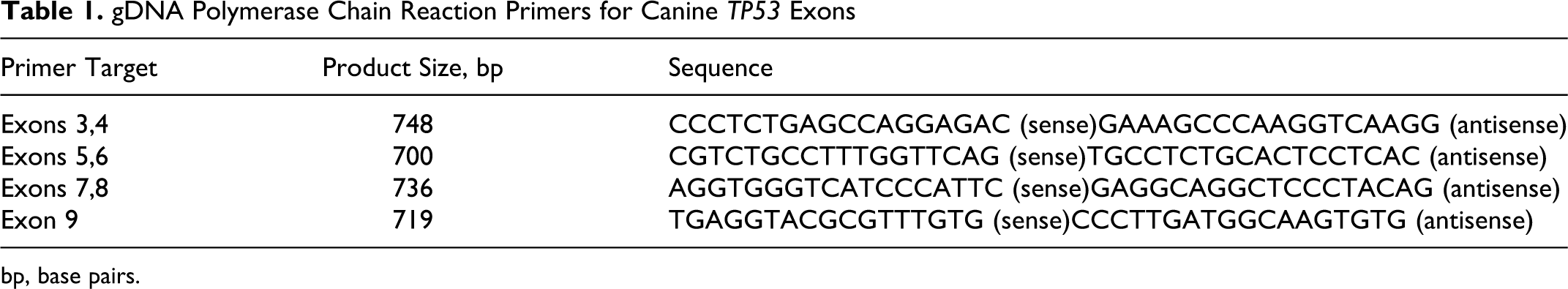
bp, base pairs.
Sequencing
Unincorporated primers and dNTPs were removed from PCR products using ExoSAP-IT (USB, Cleveland, Ohio) according to the manufacturer’s instructions. Sequencing reactions were done using the Big Dye Terminator Cycle Sequencing Kit 3.1 (Applied Biosystems, Foster City, California). Primers for exons 3, 4, and 9 were reoptimized to position primers downstream of poly-C and poly-A sequences in intron 2 and intron 8, respectively (Table 2). Two separate sequencing reactions were carried out (1 forward, 1 reverse) for each of the 4 exon-specific products. Reactions were carried out in 15-μl volumes containing 2 μl of Big Dye Terminator, 2 μL of Big Dye 5× Buffer, 1 μL of 4μM primer, and 10 to 20 ng of PCR product. Sequencing products were cleaned over Centri-Sep Spin Columns (Princeton Separations, Adelphia, New Jersey) and separated on an ABI 3730 DNA Analyzer (Applied Biosystems). Sequences were visualized using the Sequencher Software (Gene Codes Corp, Ann Arbor, Michigan) and sequence identity confirmed with a BLAST search. All ambiguous sequences or sequences with mutations were repeated.
Primers for Big Dye Sequencing Reactions

P53 cDNA Analysis
Total RNA was extracted from tumor samples using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA integrity, concentration, and purity were assessed by agarose gel electrophoresis and spectrophotometry. RNA was used to produce cDNA in a standard reverse transcription reaction. Briefly, 2 μg of total RNA was mixed with 50 ng of random primers and 5 μl of a 20mM dNTP mix (5mM for each dNTP, Thermo Fisher Scientific) for a total volume of 35.5 μl. This solution incubated at 65°C for 5 minutes, after which the reaction volume was increased to 50 μl by adding 5mM DTT, reaction buffer, 1 U of RNAseOUT, and 4 U of Superscript III Reverse Transcriptase (Invitrogen). The mixture was incubated for 1 hour at 50°C, followed by 15 minutes at 70°C. cDNA was precipitated overnight with ethanol and sodium acetate at –20°C and reconstituted in 5 μl of TE and 35 μl of sterile water. Primers for p53 spanning exons 7–11 were designed as above (Table 3), and PCR reactions were done similar to gDNA with an annealing temperature of 62°C and 32 cycles.
cDNA Primers for Canine p53
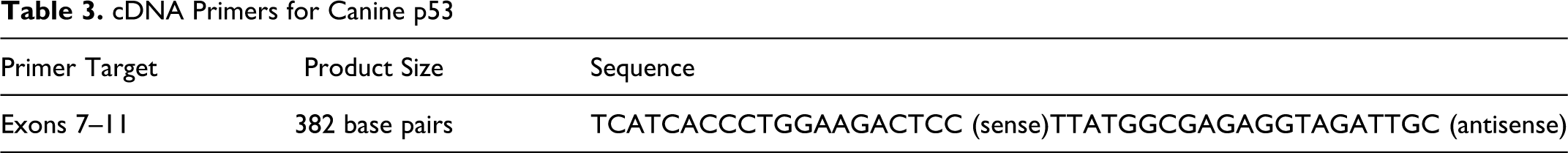
Results
gDNA was extracted from 88 tumors: 41 samples from necropsy, 10 from stereotactic computed tomography–guided biopsies, 37 from surgical resections. Tumor types were as follows: astrocytoma, 18 (grade I–pilocytic, 1; grade II, 9; grade III, 1; grade IV–GBM, 7) oligodendroglioma, 14 (grade III, 11; undetermined grade, 3) mixed glioma, 4 (grade II, 1; grade III, 2; grade IV, 1) uncharacterized glioma, 2 choroid plexus tumor, 13 (grade I–papilloma, 4; grade III–carcinoma, 9) ependymoma, 2 (grade II) histiocytic sarcoma, 3 (primary central nervous system, 2; undetermined, 1) meningioma, 32 (grade I, 18; grade II, 12; grade III, 1; undetermined grade, 1)
Control-matched gDNA was available for 64 of 88 tumor samples. Dogs’ ages ranged from 2 to 16 years, with 47 male and 41 female animals. Previously described mutations in the 3 positive control canine mammary tumor cell lines (CMT3, CMT7, CMT8) were confirmed using the described protocol (Table 4). No mutations were found in the 4 canine glioma cell lines (J3T, J3T-Bg, SDT-3, G06A).
TP53 Gene Mutations a
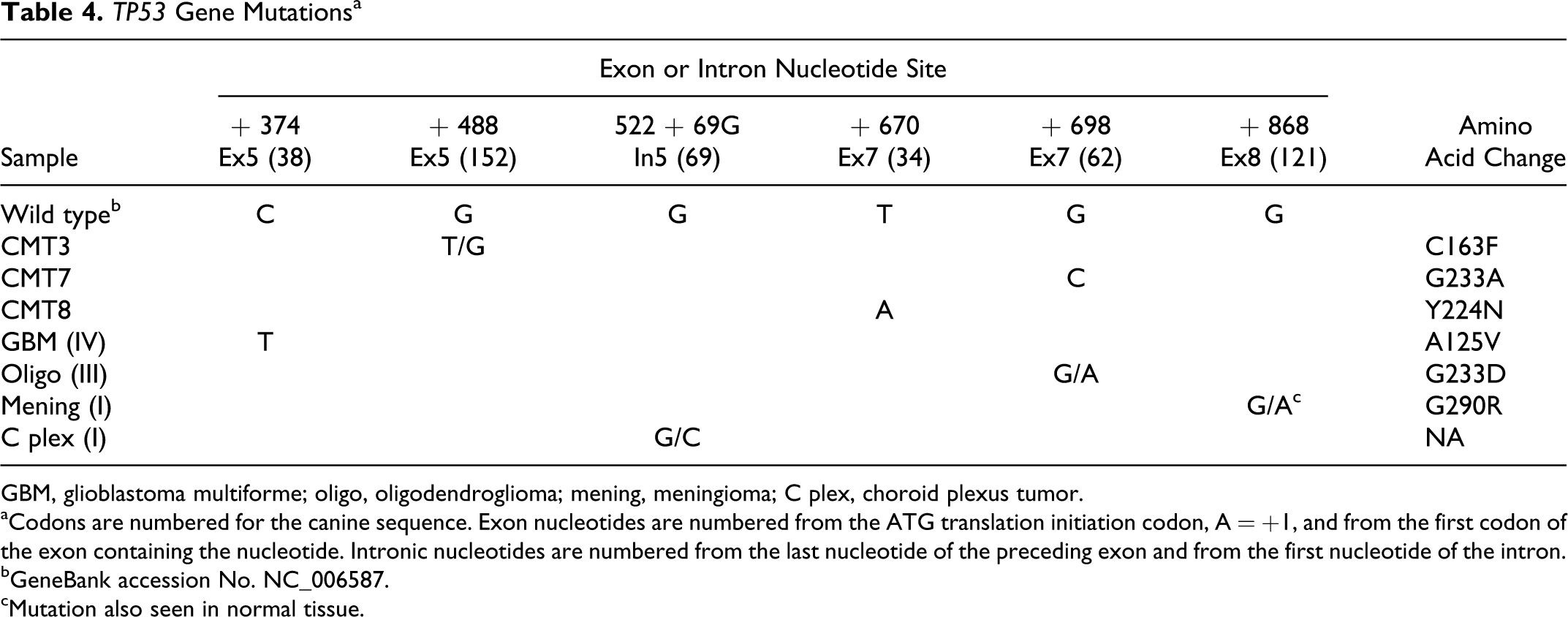
GBM, glioblastoma multiforme; oligo, oligodendroglioma; mening, meningioma; C plex, choroid plexus tumor.
aCodons are numbered for the canine sequence. Exon nucleotides are numbered from the ATG translation initiation codon, A = +1, and from the first codon of the exon containing the nucleotide. Intronic nucleotides are numbered from the last nucleotide of the preceding exon and from the first nucleotide of the intron.
bGeneBank accession No. NC_006587.
cMutation also seen in normal tissue.
Three separate exonic mutations were found in exons 5, 7, and 8 of 3 different tumor samples, resulting in an overall exonic mutation frequency of 3.4% (Table 4).
Astrocytoma IV–GBM
A C-to-T mutation with no wild-type allele was found at nucleotide position 38 of exon 5. The mutation results in the amino acid substitution of alanine with valine at codon 125 (homologous to human codon 138). The mutation was not present in autologous muscle tissue. Exonic mutation frequency in astrocytomas was 5.5% (4.5% if mixed gliomas were included).
Oligodendroglioma III
A G-to-A mutation with the wild-type allele was found at nucleotide position 62 of exon 7. The mutation results in the amino acid substitution of glycine with aspartic acid at codon 233 (homologous to human codon 245). The mutation was not present in autologous muscle tissue. Exonic mutation frequency in oligodendrogliomas was 7.1%. The overall mutation frequency in all gliomas combined (astrocytoma, oligodendroglioma, mixed/uncharacterized glioma) was 5.3%.
Meningioma I–meningothelial
A G-to-A mutation with the wild-type allele was found at nucleotide position 121 of exon 8. The mutation results in the amino acid substitution of glycine with arginine at codon 290 (homologous to human codon 302). Autologous muscle tissue was also heterozygous for the mutation. Exonic mutation frequency in meningiomas was 3.1%.
Choroid plexus tumor I
A G-to-C mutation with the wild-type allele was found at position 69 of intron 5. Corresponding nontumor tissue was not available for sequence comparison.
Preliminary sequence data identified a dinucleotide CT insertion between nucleotide 109 and 114 of intron 9 creating a CTCTCT motif in 68 of 88 tumors. Of the 68 samples with insertions, 38 were heterozygous. Intron 9 sequences from 4 control muscle samples, corresponding to 1 GBM, 1 oligodendroglioma (same oligodendroglioma sample in which the exon 7 mutation was found), and 2 choroid plexus carcinomas, were evaluated for the presence of the insertion and compared to their corresponding brain tumor sequences. A difference was found in one of the choroid plexus samples where no insertion was seen in the tumor sample, while the muscle sample was heterozygous for the insertion. No correlation was found between this CT insertion and tumor pathology or signalment.
mRNA was extracted from 12 tumor samples (3 heterozygous for the intron 9 insertion, 6 homozygous for the insertion, and 3 with no insertion). Amplification of cDNA product spanning exons 7–11 produced a single band at the expected 382 base pair size in all samples.
Discussion
Exonic mutations of the TP53 gene in canine primary brain tumors appear to be infrequent compared to human tumor counterparts based on the current data. Overall, < 4% of the canine brain tumors had exonic TP53 mutations (< 3% had mutations that were likely to result in functional alterations), compared to approximately 26% of human brain tumors. 30 TP53 mutations occur most frequently in human astrocytic tumors, although frequency varies considerably depending on specific subtype and grade. Although relative frequency of grade and subtype varies between human and canine astrocytic tumors, the decreased overall frequency of mutations in canine astrocytic tumors as a group of 4.5% (1 of 22; grades I–IV and mixed tumors) compared to about 40% in human tumors (IARC TP53 Database) 30 is a noteworthy finding. It is also consistent with the reported low frequency of mutations (1 of 12) in a previous study in dogs. 36 The low TP53 mutation frequency seen in non-astrocytic canine brain tumors (< 8%) was more consistent with reports in human tumor counterparts.
Over 90% of human TP53 tumor mutations cluster in the DNA-binding domain (exons 4–8) covered in the current study. Based on reported human brain tumor mutations, < 1% of mutations should have been missed by sequencing canine exons 3–9 (IARC TP53 Database). 30 This assumes that region- and site-specific mutation frequency is similar in dogs and humans. Certainly, the majority of canine mutations have been reported in the DNA-binding domain; however, this frequency may be overestimated, since almost all investigators have limited their analysis to this region. Some early and RNA-based studies looked at the complete coding region, 7,34,39 and one study looked at the promoter region. 22 In these cases, and consistent with human data, mutations outside the well-described predilection sites were not found or were uncommon. Approximately 25% of all TP53 mutations in human tumors occur at so-called hot spots, specifically human codons 175, 245, 248, 249, and 273 within the critical DNA-binding domain of the p53 protein. The most commonly reported mutated codons in human brain tumors are 273, 248, 175, 282 and 245 in decreasing frequency (IARC TP53 Database). 30 The most frequent TP53 alterations in human brain tumors are GC–AT mutations, with approximately 70% associated with CpG sites. 4,30 All the exonic mutations in the current study were also GC–AT alterations; only the GBM mutation, however, involved a CpG site. The exon 7 mutation in the oligodendroglioma (canine codon 233) corresponding to human codon 245 was the only mutation associated with a classic hot spot and has been reported in human GBMs. This G245D mutation has been shown to have an absence of normal p53 function, as well as inhibition of wild-type p53 activity (dominant negative), 8,15,30 so that significant loss of p53 function in this tumor was likely despite the presence of wild-type protein. The GBM exon 5 mutation in codon 125 (no wild-type allele) is equivalent to the human mutant A138V reported in a human anaplastic astrocytoma and shown to have reduced p53 function, dominant negative activity, and the ability to induce astrocytomas in experimental mice. 12,15,30 The exon 8 mutation in the meningioma, equivalent to human mutant G302A, has been reported in only 1 nonbrain tumor (tongue squamous cell carcinoma) and is of questionable significance, since it was a germ-line mutation that has minimal predicted effect on protein structure and function and has been shown to result in normal transcriptional activation. 15,30 This is consistent with its location outside the critical human DNA-binding domain codons 96–286 and tetramerization domain (328–347). 15 The intron 5 mutation in the choroid plexus carcinoma is of unknown significance and did not involve known splice sites. Approximately 0.7% of all reported TP53 mutations in human brain tumors are intronic; 30 however, unless they affect constitutive splice sites, they are most often considered benign polymorphisms. Increased incidence of a TP53 intron 6 mutation (G13964C) with significant activity has been reported, 18 although the significance has been disputed. 20
Alternative splicing of genes in normal tissues is often accomplished by exon skipping. Although control of alternative splicing is poorly understood, several regulatory proteins have been identified that bind to sequence elements and affect spliceosome assembly. Unnatural exon skipping has been reported in several cancers and inherited diseases due to gene mutations. Most often this occurs in exons or at constitutive splice sites; however, intronic regulatory elements may also be involved. Polypyrimidine tract binding protein (PTB) binds to pyrimidine-rich intronic elements and has been associated with inhibition of exon splicing in several genes. 5 CU dinucleotide repeats serve as a binding site for the PTB family members, and pentameric and hexameric sequence motifs (CTCTC, CTCTCT) have been reported as splicing signals in exon flanks. 5,14,46 The intron 9 insertion observed in 68 of 88 tumor samples would create a putative PTB binding site (CTCTCT), that might result in unnatural exon skipping. However, analysis of cDNA from tumor samples spanning exons 7–11 revealed no evidence of truncated or alternative transcripts, suggesting that the insertion represents a nonfunctional polymorphism.
Direct sequencing is the IARC-recommended method for determining TP53 mutational status. Indirect methods, such as immunohistochemistry, may provide additional information regarding aberrant processing of p53 protein; however, it cannot distinguish among different types of mutations, does not detect truncating mutations, can be variable depending on operator and antibody used, and can give false-positive results. 30 Intronic primer pairs were used to amplify the TP53 exonic sequence to minimize the potential for amplification of TP53 pseudogenes.
Given the key role of the p53 pathway in oncogenesis in a variety of tumor types, it is likely that aberrant p53 function is also involved in canine brain tumors despite the low frequency of p53 mutations. Additional mechanisms of p53 pathway inactivation have been well described, including overexpression of p53 inhibitors such as MDM2, 3,27,29,32 inactivation by protein binding or cytoplasmic retention, 26 and defects in upstream or downstream effectors. 1,31 Loss of heterozygosity may also play a role in decreased p53 function, and the apparent loss of the intron 9 CT insertion in one of the choroid plexus tumors suggests that this might have occurred, although gene conversion cannot be excluded. It is also possible that the p53 pathway does not play a key role in canine brain tumor formation and progression; however, p53 pathway abnormalities are considered as obligate events in the pathogenesis of human high-grade gliomas. 6 Therapeutic strategies targeting the p53 pathway are currently in development, and clinical trials have been reported including MDM2 inhibitors and TP53 gene transfer (reviewed in Lane et al 17 ). Interrogation of canine tumor–derived cell lines may provide a platform for detailed assessment of p53 function and response to targeted therapy, since appropriate selection of strategies based on the specific underlying tumor pathology is likely to be an important factor in efficacy. Absence of TP53 gene mutations in canine brain tumors suggests that more detailed studies are warranted investigating alternative genetic and epigenetic mechanisms of p53 pathway perturbation to define the presence and character of such events.
Footnotes
Notes
Acknowledgements
This research was supported by the Paul C. and Borghild T. Petersen Foundation and the Student Training in Advanced Research Program, University of California, Davis (A.N.W.).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research was supported by the Paul C. and Borghild T. Petersen Foundation.