
Abstract
There is evidence that genetic factors play a role in the complex multifactorial pathogenesis of hydrocephalus. Identification of the genes involved in the development of this neurologic disorder in animal models may elucidate factors responsible for the excessive accumulation of cerebrospinal fluid in hydrocephalic humans. The authors report here a brief summary of findings from 12 lines of genetically engineered mice that presented with autosomal recessive congenital hydrocephalus. This study illustrates the value of knockout mice in identifying genetic factors involved in the development of congenital hydrocephalus. Findings suggest that dysfunctional motile cilia represent the underlying pathogenetic mechanism in 8 of the 12 lines (Ulk4, Nme5, Nme7, Kif27, Stk36, Dpcd, Ak7, and Ak8). The likely underlying cause in the remaining 4 lines (RIKEN 4930444A02, Celsr2, Mboat7, and transgenic FZD3) was not determined, but it is possible that some of these could also have ciliary defects. For example, the cerebellar malformations observed in RIKEN 4930444A02 knockout mice show similarities to a number of developmental disorders, such as Joubert, Meckel-Gruber, and Bardet-Biedl syndromes, which involve mutations in cilia-related genes. Even though the direct relevance of mouse models to hydrocephalus in humans remains uncertain, the high prevalence of familial patterns of inheritance for congenital hydrocephalus in humans suggests that identification of genes responsible for development of hydrocephalus in mice may lead to the identification of homologous modifier genes and susceptibility alleles in humans. Also, characterization of mouse models can enhance understanding of important cell signaling and developmental pathways involved in the pathogenesis of hydrocephalus.
Hydrocephalus is characterized by the excessive accumulation of cerebrospinal fluid (CSF) within brain ventricles, which results in ventricular dilatation and damage to the surrounding brain parenchyma. CSF is primarily produced at the choroid plexus, and it flows from the lateral ventricles to the third ventricle via narrow passageways and then to the fourth ventricle via the aqueduct of Sylvius. From the fourth ventricle, CSF passes through foramina into the subarachnoid space where it is believed to be drained via nasal lymphatics or absorbed through the arachnoid villi. 54,104 The circulation of CSF within the central nervous system (CNS) is aided by the pulsations of the choroid plexus and by movement of motile cilia on ependymal cells. Hydrocephalus can be caused by blockage of aqueducts connecting the brain ventricles, by impaired flow of CSF, by reduced CSF absorption, or even by excessive CSF production. 21 Hydrocephalus resulting from an obstruction in CSF flow along one or more of the narrow passages connecting the ventricles is classified as noncommunicating hydrocephalus, whereas cases resulting from impaired absorption of CSF in the subarachnoid space are termed communicating hydrocephalus. 76 Abnormalities of the cerebral aqueduct or subarachnoid space were observed in a majority of cases of hereditary hydrocephalus in humans, 11 with impaired absorption in the subarachnoid space or overproduction at the choroid plexus being less commonly implicated. 72 Left untreated, all forms of hydrocephalus can result in progressive enlargement of the head, convulsions, mental disability, and death.
Hydrocephalus affects 1 to 3 of 1000 children at birth and is the most common neurologic disorder requiring surgery in children. 13 Congenital hydrocephalus may be caused by either genetic or environmental factors, which include brain malformations, intracerebral hemorrhage, maternal alcohol use, infections, or X-ray radiation during pregnancy. 111 There is growing evidence that genetic factors can be involved in the pathogenesis of hydrocephalus in humans, 77,87,92 and an estimated 40% of human cases have a genetic component. 42 However, to date, mutations in only 1 human gene (L1CAM) have been definitively linked to the development of congenital hydrocephalus, and these cases account for only 5% to 15% of likely hereditary cases. 58 The identification of the specific genes responsible for the development of congenital hydrocephalus in humans is complicated by the likely involvement of multiple genes and genetic background effects.
In mice, congenital hydrocephalus is a complex polygenic trait, and its development is strongly influenced by the presence of strain-associated genetic modifiers. It has long been recognized that hydrocephalus is more common in C57BL strains than in other common inbred strains of mice. 89 Differences in genetic background can have a large impact on experimental studies. For example, nm1054- or fyn-deficient mice on C57BL/6 backgrounds develop severe hydrocephalus, whereas mutants on a 129 or mixed background showed either mild or no hydrocephalus. 38,61 In L1-deficient mice (with L1 being a neural cell adhesion protein encoded by L1CAM), severe hydrocephalus was seen only after backcrossing to the C57BL/6 strain, with the mutation becoming embryonic lethal after several generations. 48 Similarly, hydrocephalus in Naglu mutant mice was at least partially related to the background strain C57BL/6. 36 It is therefore likely that our findings were influenced by the mixed genetic background of the B6129F1 hybrid mice used in these studies.
In generating and analyzing over 4650 knockout mouse lines in a high-throughput mutagenesis and phenotyping process designed to characterize protein functions and identify novel drug targets, 108,109 we have discovered many phenotypes in mice that may prove useful in elucidating fundamental processes in biology and the pathogenesis of disease. We report here a summary of findings from 12 lines of genetically engineered mice that presented with autosomal recessive congenital hydrocephalus. This study illustrates the value of using knockout mice to identify genetic factors involved in the development of congenital hydrocephalus. Although we did not elucidate the precise mechanisms involved in the pathogenesis of hydrocephalus in our studies, our findings suggest that dysfunction of motile cilia may be the most common underlying mechanism responsible for congenital hydrocephalus in mice.
Materials and Methods
Mouse Production
For all lines included in this report, the complete allele symbols are provided in Tables 1–3. For brevity, only the basic gene symbols are used in the text. For those knockout lines generated by gene trapping, we used strain 129S5SvEvBrd–derived embryonic stem (ES) obtained from the OmniBank library as described previously. 108 Intragenic gene trap mutations were selected from the OmniBank library by identifying the precise location of vector insertion in each clone of interest using inverse genomic polymerase chain reaction. 1 Briefly, oligonucleotide primers complementary to the gene trap vector were used to amplify the vector insertion site for each clone, which was then compared to mouse genome sequence assemblies to localize the insertion with respect to the exons and introns of the gene (Table 1). Gene disruption in vivo was confirmed by a direct analysis of gene expression using reverse transcription PCR (RT-PCR). For this, RNA was extracted from at least 2 tissues of wild-type and mutant mice using a bead homogenizer and RNAzol (Ambion, Austin, TX) according to manufacturer’s instructions. Reverse transcription was performed with SuperScript II (Invitrogen, Carlsbad, CA) and random hexamer primers, according to the manufacturer’s instructions, with PCR amplification being performed with oligonucleotide primers complementary to exons flanking the insertion site.
Genome5000 Knockouts Exhibiting Hydrocephalus
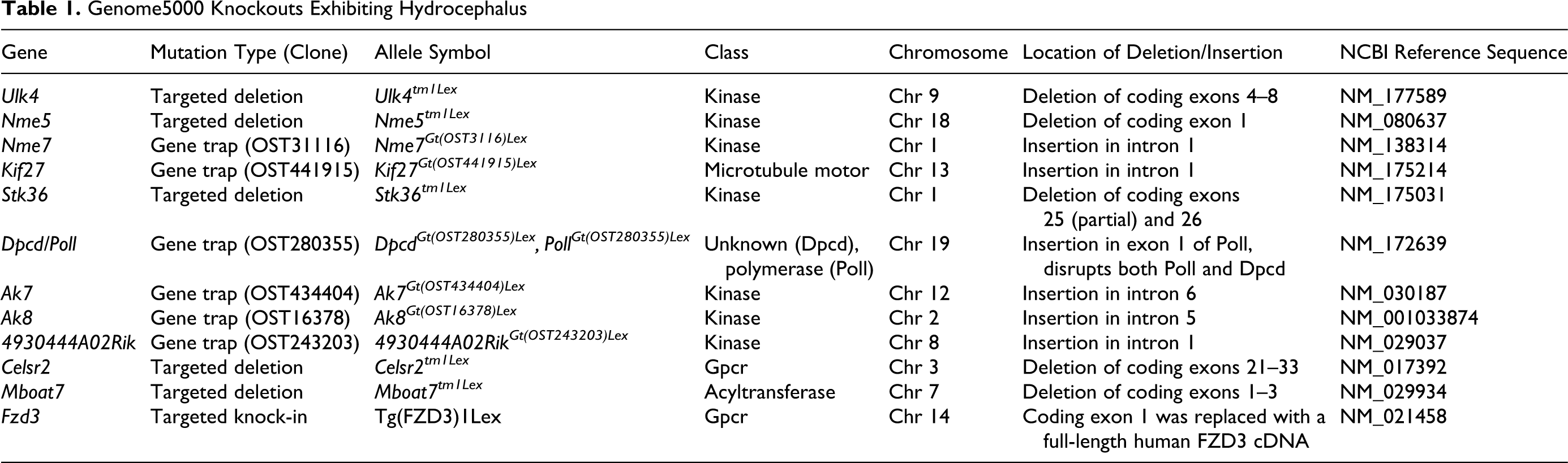
Genome5000 Knockouts Exhibiting Hydrocephalus: Nucleotide Sequences

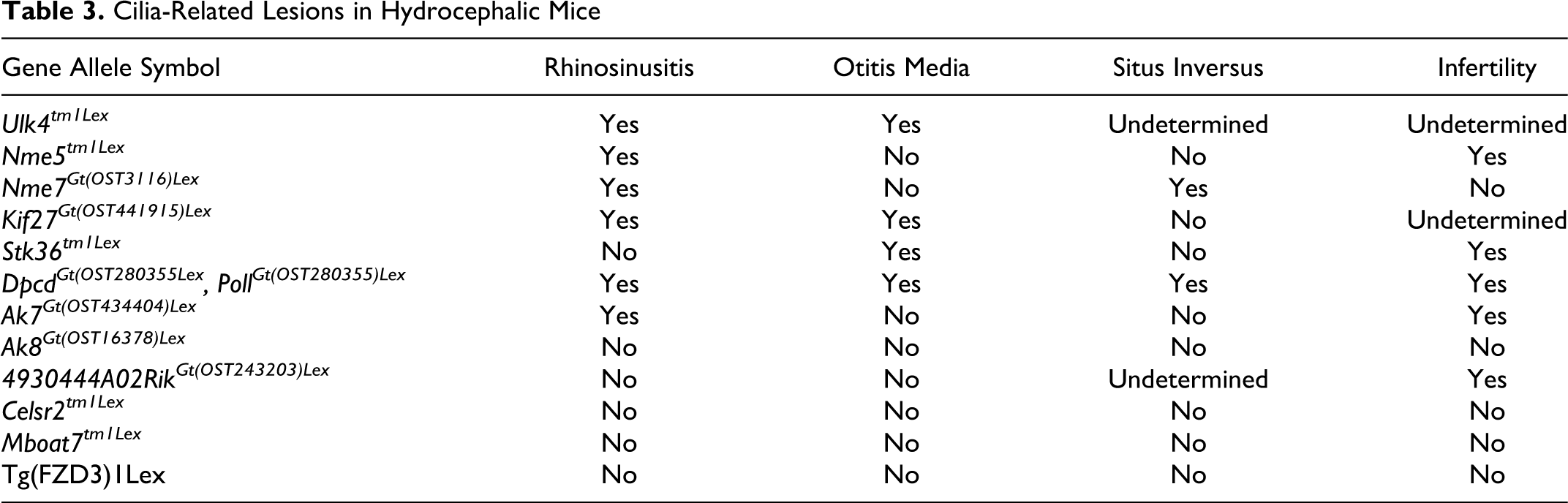
Cilia-Related Lesions in Hydrocephalic Mice
For those lines generated by homologous recombination, exons of interest were deleted and replaced with a selection cassette containing the neomycin gene. To generate pKOS targeting vectors, a lambda KOS phage library 103 was screened by duplex PCR using oligonucleotide primers complementary to the gene of interest. The genomic clone identified in this screen was cotransformed into yeast with a URA3 yeast selection cassette that had been appended with gene-specific arms flanking the desired deletion interval (Table 1). Clones that had undergone homologous recombination with the yeast-selectable marker were isolated, and the yeast cassette was replaced with an ES cell selection cassette containing the reporter gene LacZ and the neomycin gene. For pKO Scrambler targeting vectors, the 5′ and 3′ arms of homology were generated by long-range PCR, and the targeting vector was generated in a single unidirectional cloning step in which 2 homology arms and the ES cell selection cassette were ligated into the pKO Scrambler NTKV-1901 vector (Stratagene, La Jolla, CA). Completed pKOS or pKO Scrambler targeting vectors were electroporated into 129S5/SvEvBrd ES cells, and G418/FIAU-resistant ES cell clones were isolated. Correctly targeted clones were identified and confirmed by Southern analysis (data not shown).
Targeted and gene-trapped ES cell clones were microinjected into C57BL/6-Tyrc-Brd (albino) blastocysts to generate chimeric animals that were bred to C57BL/6-Tyrc-Brd (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous gene-deficient mice. Knockout F2 mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (–/+) offspring of chimeric founder parents. Therefore, all mice used in these studies are of mixed C57BL and 129 genetic background. In all studies reported here, mutant mice were compared directly with their wild-type littermates used as negative controls. Using the albino variant of C57BL/6 mice (C57BL/6-Tyrc-Brd) permits simple visual recognition of chimeric offspring because they have dark eyes and patches of dark hair that derive from stem cells from the agouti 129S5/SvEvBrd. Genotypes of offspring were determined by quantitative PCR as previously described. 41 In brief, DNA isolated from tail biopsy samples was assayed by quantitative PCR for the neo gene, which is present in both the gene-targeting and gene-trapping vectors used to generate the mutations described in this study. In the case of Fzd3, a humanized knock-in transgenic mouse designated as Tg(FZD3)1Lex was made by replacing the first coding exon of mouse Fzd3 with a selection cassette containing a full-length cDNA for human FZD3. Expression of the human FZD3 gene in knock-in mice was confirmed by RT-PCR analysis of multiple tissues from heterozygous Tg(FZD3)1Lex mice.
Mouse Husbandry
Mice were housed in microisolator cages within a barrier facility at 24°C and on a fixed 12-hour-light/12-hour-dark cycle and were provided ad libitum acidified water and Purina rodent chow No. 5001 (Purina, St Louis, MO). Procedures involving animals were conducted in conformance with Lexicon’s Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the National Research Council’s Guide for the Care and Use of Laboratory Animals (1996). Quarterly sentinel surveillance showed no evidence of pathogenic rodent viruses, Mycoplasma, or Helicobacter spp. in the Lexicon Pharmaceutical source colonies.
Histopathology
Immediately after euthanasia, knockout mice and age-matched normal control mice were routinely fixed by cardiac perfusion with 10% neutral buffered formalin. Tissues were collected and immersed 10% neutral buffered formalin for an additional 48 hours except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin for histopathologic examination.
β-Galactosidase Staining
β-Galactosidase (β-Gal) activity was assayed as previously described. 98 Briefly, anesthetized mice were perfused sequentially with β-Gal fixative (0.2% glutaraldehyde, 1.5% paraformaldehyde, 2mM MgCl2, 5mM EGTA [ethylene glycol], 100mM sodium phosphate [pH 7.3]), followed by 2 ml of β-Gal rinse (0.2% Nonidet-P40, 0.1% sodium deoxycholate, 2mM MgCl2, 100mM sodium phosphate), and, finally, 10 ml of β-Gal stain (5mM K3Fe(CN)6 [potassium ferricyanide], 5mM K4Fe(CN)6 [potassium ferrocyanide], 1 mg/ml of 5-bromo-4-chloro-3-indolyl-D galactopyranoside [β-Gal; dissolved in dimethylformamide], 0.2% Nonidet-P40, 0.1% sodium deoxycholate, 2mM MgCl2, 100mM sodium phosphate [pH 7.3]). Tissues were postfixed in β-Gal fix, rinsed in the β-Gal rinse, and then incubated in β-Gal stain solution for 48 hours. After 3 additional washes in β-Gal rinse, the tissues were postfixed in Bouin’s fixative before dehydration and embedding in paraffin. Sections were cut at 4 μm and counterstained with nuclear fast red.
Results
Ulk4 (unc-51-like kinase 4 [C. elegans]): 28 Grossly, all mutant mice were smaller than sex-matched littermates, and all exhibited domed heads typical of hydrocephalus. The majority of the mutant mice died before reaching 4 months of age. Histologically, marked to severe dilatation of the lateral and third ventricles was evident in knockout mice. Hemorrhage was frequently present in the lateral and third ventricles of affected mice (Fig. 1 ), and the hemorrhage was sometimes accompanied by fibrosis and neovascularization of the meninges and choroid. Strikingly, the nasal passages and maxillary sinuses of knockout mice were partially filled with varying combinations of proteinaceous fluid or suppurative exudates (Fig. 2 ). Suppurative otitis media was also present in many animals. Although cilia were seen histologically on respiratory epithelium and ependymal cells lining the dilated ventricles, they appeared to be shorter than those in wild-type littermate control mice. Because of early mortality, spermatozoal flagella and male fertility could not be assessed in the knockout mice, but seminiferous tubules and spermatids in the young males examined were histologically identical to those in age-matched control littermates. Situs determination was not completed in this line.
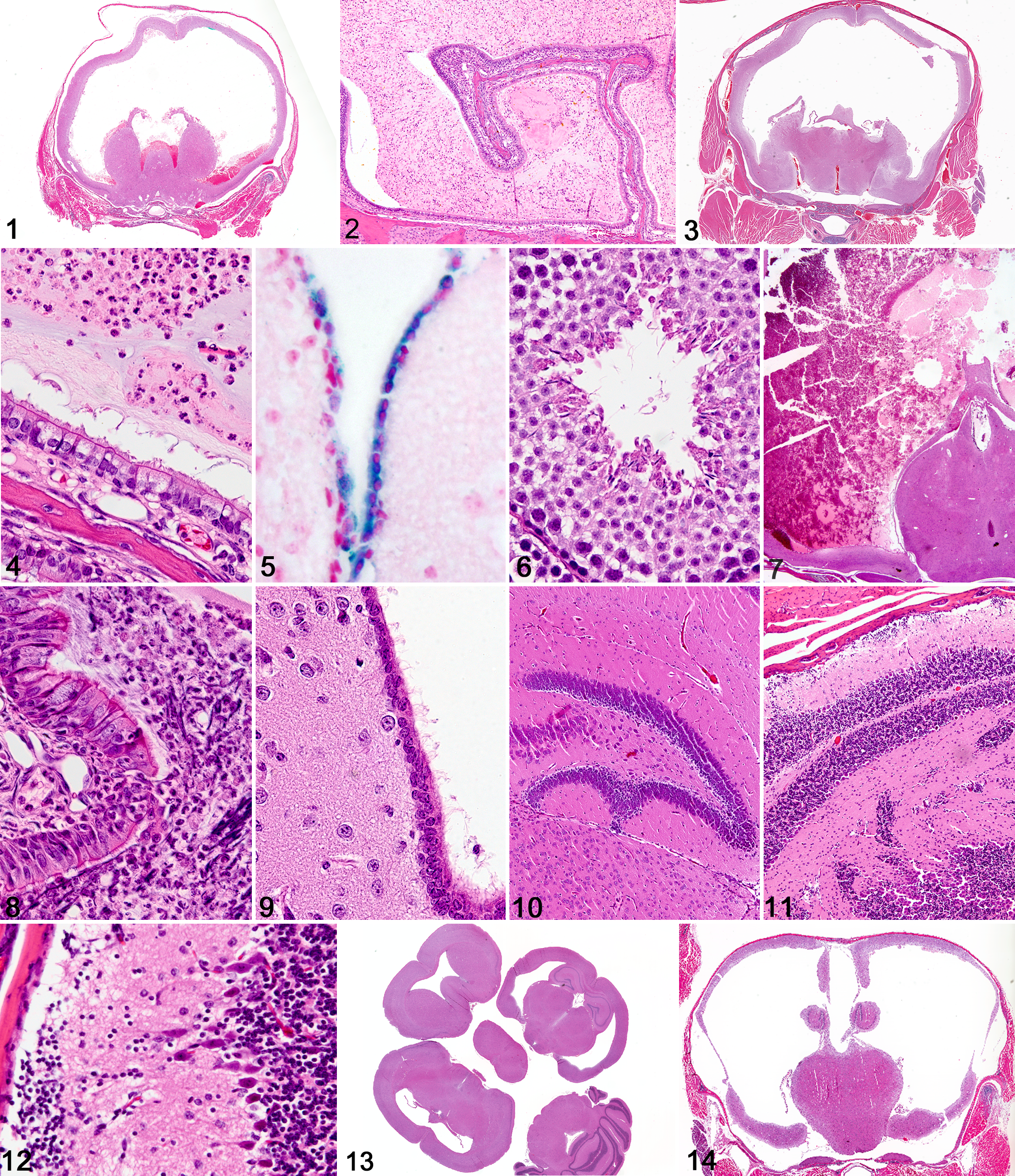
Nme5 (nonmetastatic cells 5, protein expressed in nucleoside-diphosphate kinase): 9,27 Grossly, doming of the skull was evident in all mutant mice. Histologically, there was moderate to marked hydrocephalus with mild intraventricular hemorrhage involving the lateral and third ventricles (Fig. 3 ). In several mice, hydrocephalus was accompanied by an accumulation of proteinaceous and suppurative exudates in nasal sinuses and passageways (Fig. 4 ). The cilia on respiratory epithelium and ependymal cells had a histologically normal appearance. In this line, we performed a LacZ assay to detect promoter activity and saw strong Nme5 reporter gene expression within ependymal cells (Fig. 5 ), respiratory epithelium, and late-stage spermatids. In several 6-week-old male mice, late-stage spermatogenic arrest and degeneration were characterized by limited formation of tails, although there was normal condensation and flattening of spermatozoa heads. Although the fertility of knockout mice was not tested clinically, in the single adult (16 weeks old) male mutant mouse available for examination, defective spermiogenesis was characterized by degeneration of late-stage spermatids and reduced formation of flagella, most of which were detached (Fig. 6 ). Degenerating and often-tailless spermatozoa were found with macrophages and cell debris in the rete testes and proximal epididymis, but aspermia was evident in all middle and distal epididymal tubules. Notably, situs inversus was not detected in any mutant mice in this line.
Nme7 (nonmetastatic cells 7, protein expressed in nucleoside-diphosphate kinase): 9,27 We recently reported in detail the phenotype of Nme7 –/– mice, which is hydrocephalus of variable severity in almost 100% of Nme7 –/– mice. 97 In the Nme7 –/– mice, doming of skulls was either undetectable or very mild, and the knockout mice demonstrated normal behavior in neurologic testing (data not shown). In addition, male and female Nme7 –/– mice were fertile when bred to wild-type mates, and most mutant mice lacked evidence of ciliary dysfunction, such as proteinaceous exudates and suppurative inflammation, in nasal passages and sinuses. Situs inversus was present in approximately 50% of mutant mice.
Kif27 (kinesin family member 27): 53 Mutant mice were small and sickly, with all mutants either dying or being euthanized by 8 weeks of age. All mutant mice available for analysis exhibited marked to severe hydrocephalus. Hemorrhage was frequently present in the lateral and third ventricles of affected mice (Fig. 7 ), and the hemorrhage was sometimes accompanied by fibrosis and neovascularization of the meninges and choroid. In all mutant mice examined, severe suppurative exudates frequently filled most of the luminal space within nasal passages and sinuses (Fig. 8 ). In some cases, there was also bilateral suppurative otitis media. Histologically, respiratory and ependymal cilia (Fig. 9 ) were indistinguishable from those of wild-type littermates. Fertility was not assessed because of early mortality, and the mutant male mice evaluated in pathology were too young to permit reliable morphologic evaluation of spermatogenesis. Situs inversus was not observed in this line.
Stk36 (serine/threonine kinase 36; fused homolog, Drosophila): 65,106 Due to ill health, the majority of the mutants were euthanized between 3 and 6 weeks of age, although several survived up to 12 weeks. All mutant mice exhibited hydrocephalus, craniofacial abnormalities, and growth retardation (reduced body weight and mean body length in males and females). We noted, other than severe hydrocephalus, bilateral suppurative inflammation in the middle ears of 3 of 5 mutants, which extended into the inner ear, meninges, and ventricles in a single mutant mouse. The 2 female Stk36 –/– mice tested were sterile. Due to the reduced viability of the mutants, no male Stk36 –/– mice were available for fertility analysis. Situs inversus was not detected in any mutant mice in this line.
Dpcd (deleted in primary ciliary dyskinesia): 110 We recently reported the occurrence of situs inversus and hydrocephalus in compound mutant Dpcd/Poll –/– mice, 97 confirming a similar phenotype reported in single mutant Dpcd –/– by another group. 56 In both studies, Dpcd –/– mice were smaller than wild-type littermates, and all mutant mice showed hydrocephalus accompanied by histologic lesions consistent with underlying ciliary dysfunction. Nasal passages and sinuses were often filled with abundant mucopurulent exudate, and otitis media was present in most animals. There was doming of the skull owing to moderate to marked dilation of the lateral and third ventricles of the brain. Males were clinically infertile, even though histologic examinination showed apparently normal spermatogenesis in seminiferous tubules and epididymides. However, examination of live spermatozoa showed that the spermatozoa were immotile, with many appearing to have shorter-than-normal tails and rounded heads. Situs inversus was present in approximately 50% of mutant mice.
Ak7 (adenylate kinase 7): 30,66,71 Our findings were essentially identical to those recently reported by another group. 30 Grossly, all Ak7 –/– mutant mice exhibited domed heads and hydrocephalus. Histologically, the most notable lesions included moderate to severe hydrocephalus, which was accompanied by chronic active rhinosinusitis characterized by accumulation of proteinaceous and mucopurulent exudates in nasal passages and paranasal sinuses in all mutant mice. Although most mutant mice died before 8 weeks of age, the few surviving mutant male mice were clinically sterile and showed histologic evidence of late-stage spermatogenic arrest and severe hypospermia. Situs inversus was not detected in Ak7 –/– mice.
Ak8 (adenylate kinase 8): 71 All homozygous Ak8 –/– mice in this line developed mild to moderate hydrocephalus, usually restricted to the lateral ventricles (Fig. 13 ). Notably, doming of the head was not observed grossly, most likely because of the later onset and milder severity of hydrocephalus. Mutant mice were viable and displayed normal behavior and growth, despite the onset of hydrocephalus. A time course study showed that hydrocephalus was not present in embryos and neonates but became evident at approximately 2 weeks of age and reached maximum severity at time of weaning (3 weeks). In most Ak8 –/– mice, the hydrocephalus was characterized by bilateral dilatation of the lateral ventricles and reduced size of the hippocampus. A surprising finding was the lack of any notable behavioral phenotype in these mice in the trace aversive conditioning test for learning and memory, despite the reduced size of the hippocampus in many hydrocephalic mice. No nasal exudates were present, and normal male and female fertility was observed in mutant mice. Situs inversus was not observed in any Ak8 –/– mice.
RIKEN cDNA 4930444A02: 12 Grossly, all mutant mice exhibited dome-shaped heads of varying severity, and most were either euthanized immediately or sent to necropsy to confirm an initial diagnosis of hydrocephalus. Surviving mutant mice that underwent neurologic testing exhibited numerous behavioral abnormalities when compared with their wild-type littermates. Mutant mice displayed tremors, and inverted screen testing showed 5 of 8 falling off, while none of the wild-type littermates fell, suggesting impaired motor strength. Impaired sensorimotor gating/attention in mutant mice was suggested by decreased prepulse inhibition, and impaired learning/memory was detected with trace aversive conditioning testing. In testing nociception, decreased paw flinching was observed during both formalin phases, suggesting decreased sensitivity to acute and tonic pain. Histologically, the most obvious changes observed in mutant mice submitted to necropsy were hydrocephalus in 4 of 5 and cerebellar dysplasia in all 5. Abnormalities in neuronal migration were evident in other parts of the brain; in the cerebral cortex, there was disorganization of cortical neuron layers, and the dentate gyrus of the hippocampus had a scalloped appearance (Fig. 10 ). The cerebellar dysplasia was characterized by multifocal disorganization of cerebellar cortical neurons, with clusters of external granular neurons being scattered on the surface of the cerebellum and multifocally within the molecular layer of the cerebellum (Fig. 11 ). In some regions, there was incomplete separation of cerebellar folia, and Purkinje cell neurons were occasionally found in the molecular layer (Fig. 12 ). These changes in the cerebral cortex, hippocampus, and cerebellum were present even in the 1 knockout mouse examined that did not have lesions of hydrocephalus. Only 1 male mouse was available for fertility testing, and no pups were produced after 60 days of breeding with 4 fertile wild-type female mice. Other than mild hydrocephalus and cerebellar dysplasia, this mutant male mouse appeared healthy, and normal appearing sperm were present in testes and epididymis. The absence of vaginal plugs in the female cage mates suggested that the infertility might be a result of abnormal motor control or mating behavior in the mutant male mouse. Situs was not evaluated in this knockout line.
Celsr2 (cadherin, EGF LAG seven-pass G-type receptor 2; flamingo homolog, Drosophila): 33,34,44,93 Grossly, Celsr2 –/– mice were produced according to normal Mendelian ratios and were indistinguishable from wild-type littermates. However, 12 of 13 mutant mice examined histologically exhibited mild to moderate dilation of the lateral and third ventricles with mild compression of the surrounding neuropil. No other lesions were observed, and the mutant mice showed normal behavior and fertility. Situs inversus was not observed in any Celsr2 –/– mice.
Mboat7 (membrane bound O-acyltransferase domain containing 7): 35 Grossly, Mboat7 –/– mice were smaller than littermate controls and had domed heads characteristic of hydrocephalus. Most Mboat7 –/– mice died or were euthanized by 1 to 3 months of age, and pathologic analysis confirmed a diagnosis of moderate to severe hydrocephalus affecting the lateral and third ventricles (Fig. 14 ). In many affected mice, only a thin rim of neocortex remained. Histologically, normal-appearing cilia were present on respiratory and ependymal epithelium, and there was no clinical evidence of defective ciliary function (normal fertility and nasal passageways) in Mboat7 –/– mice. Situs inversus was not observed in any Mboat7 –/– mice.
Fzd3 (frizzled homolog 3; Drosophila): 101 Fzd3 –/– mice produced by 2 strategies (data not shown) resulted in the production of 3 null mice that had curly or looped tails and died within hours of birth. In contrast, the humanized FZD3 knock-in mice designated as Tg(FZD3)1Lex (in which the targeted mouse Fzd3 was replaced with its human FZD3 ortholog) and described in this report were viable. RT-PCR analysis revealed that the human transcript was present in the humanized mutant mice. Although the homozygous Tg(FZD3)1Lex mice survived to adulthood, most of these mice exhibited varying degrees of dilatation of the lateral and third ventricles. No other notable pathology phenotypes were observed in the humanized mutant mice. Behavioral testing on a cohort of 10 homozygous Tg(FZD3)1Lex mice exhibited some neurologic changes in comparison to wild-type littermates, such as a decreased depressive-like response during tail suspension testing, impaired sensorimotor gating/attention during prepulse inhibition testing, and impaired learning/memory during trace aversive conditioning, suggesting impaired learning/memory in the transgenic mice. No notable differences were observed in tests of circadian rhythm activity, stress-induced hyperthermia, open field testing, inverted screen, hot plate, and marble burying. Histologically, normal-appearing cilia were present on respiratory and ependymal epithelium, and there was no clinical evidence of defective ciliary function (normal fertility and nasal passageways) in Tg(FZD3)1Lex mice. Situs inversus was not observed in any homozygous Tg(FZD3)1Lex mice.
Discussion
We identified 12 hydrocephalic lines in a survey of over 4650 independent knockout mouse lines, and our phenotypic findings and evaluation of available gene information suggest that dysfunctional motile cilia were the underlying pathogenetic mechanism in 8 of the 12 lines that developed autosomal recessive congenital hydrocephalus. We were unable to determine a likely underlying cause in the remaining 4 lines, but it is possible that some could have ciliary defects specific to ependymal cilia. In at least 1 case, the pathogenetic mechanism appears to involve disruptions in normal neuronal migration and/or brain morphogenesis.
The probable involvement of ciliary dysfunction in the pathogenesis of congenital hydrocephalus in 8 of the 12 mutant mouse lines reported here is not surprising given the previous frequent association of hydrocephalus with ciliary defects in mice. 8 Abnormal or deficient motility of ependymal cilia during brain morphogenesis clearly contributes to the development of hydrocephalus in mice. The synchronous beat of ependymal cilia that line the ventricles and interventricular connections generates a directional flow of CSF, which has been termed “ependymal flow.” 47 It appears that a steady ependymal flow is required to maintain the patency of the aqueduct during brain development because the absence of ependymal flow results in secondary aqueduct stenosis during early postnatal brain development. 47 Interestingly, cilia might also be involved in the regulation of CSF production. One form of communicating congenital hydrocephalus has been attributed to increased secretory activity of the choroid plexus in mice lacking the E2f5 transcription factor. 62 In addition, partly because hydrocephalus in Tg737orpk mice develops before motile cilia appear, the disorder in these mice has been attributed to overproduction of CSF by choroid plexus epithelium resulting from defects in primary cilia. 7,8
Previously reported mutant cilia-related genes in hydrocephalic mice include mdnah5 (mouse axonemal dynein heavy chain), 46,47 hy3 (hydin), 23,24 Pcdp1 (primary ciliary dyskinesia protein 1), 61 SPAG6 (sperm-associated antigen 6), 83 RFX (regulatory factor X), 5 and Tg737 (IFT88, polaris). 91 Mice deficient in axonemal dynein heavy chain 5 (Mdnah5) exhibit immotile cilia, random left-right axis specification, chronic respiratory infections, and hydrocephalus. 46 Disruption of the outer dynein arm protein in Mdnah5 –/– mice impairs ependymal cell ciliary motility, and the resulting disruption of CSF flow is believed to result in aqueduct closure and hydrocephalus during early postnatal development. 47 Similarly, mice with mutations in the ciliary protein Spag6 or hydin or the transcription factor Hfh4 lack ependymal cell cilia and exhibit hydrocephalus. 14,23,83 Congenital hydrocephalus in hy3 mice is associated with the loss of cilia from many ependymal cells and was linked to a frame shift mutation in the Hydin gene. 23 Hydin is a large protein that localizes to a single microtubule on the central apparatus of cilia/flagella and is required for motility. 59 The ciliary dyskinesis in hydin-null mice greatly reduced or eliminated the flow generated by ependymal cilia. 59 Hydrocephalus is observed in Tg737orpk mutant mice that are homozygous for a hypomorphic allele of IFT88/polaris 8 that is part of the large intraflagellar transport particle that mediates the bidirectional movement of proteins from the base of cilia to the cilia tip 43,84 required for the assembly of motile and immotile cilia. 74,91 Tg737orpk mice develop a variety of ciliopathies that suggest the involvement of both motile and immotile cilia; these include hydrocephalus, cystic kidney disease, sterility, biliary and bile duct hyperplasia in the liver, acinar cell atrophy in the pancreas, retinal degeneration, and skeletal patterning abnormalities. 112 However, it should be noted that hydrocephalus in Tg737orpk has also been attributed to defects in primary cilia that result in overproduction of CSF by choroid plexus epithelium. 8
The involvement of ciliary dyskinesia in the pathogenesis of hydrocephalus is well established in mice, but this association is less clear in humans, as most patients with primary ciliary dyskinesia do not develop hydrocephalus. Nevertheless, there is considerable evidence showing an increased incidence of hydrocephalus in human patients with primary ciliary dyskinesia 3,6,25,26,39,49 or situs inversus. 2,57 Similarly, an increased incidence of hydrocephalus has been associated with primary ciliary dyskinesia in certain dog breeds. 20,22,29,80 The mechanical function of ependymal flow would be most important in narrow passageways, therefore making the cerebral aqueduct a critical location for an intact and functional ependymal lining. Obstruction of the aqueduct would be more likely during embryonic development, when the physical size of the lumen is small. 25 However, it is possible that the shorter and wider aqueduct in humans could result in a reduced vulnerability to secondary blockage of the aqueduct. 47 The apparently increased susceptibility of mice to cilia-related hydrocephalus could be the result of specific anatomical characteristics of the mouse brain. Specifically, the relatively long and narrow aqueduct of mice might make them much more susceptible to the effects of dysfunctional ependymal cilia.
There was strong evidence supporting a role for dysfunctional motile cilia in the majority of lines described in this report, with observed phenotypes ranging from mice showing evidence of motile cilia dysfunction in multiple locations (rhinosinusitis, situs inversus, infertility, and hydrocephalus) to mice showing only hydrocephalus. There is abundant evidence that some of the proteins, developmental processes, and motility of cilia are location dependent, 32 so it is entirely understandable that the effects of specific gene mutations could be restricted to only a subset of ciliated cells. For example, it is known that ependymal cilia are longer than respiratory cilia and beat at approximately twice the frequency of respiratory cilia. 70 Therefore, it might be expected that mutations in individual cilia genes, such as those we observed in the Ak8 knockout mice, could preferentially affect cilia on ependymal cells and thereby cause tissue-specific functional deficits (hydrocephalus). Conversely, we previously found evidence that Ttll1 knockout mice have dysfunctional respiratory cilia, but the absence of hydrocephalus in those mice suggested that this mutation had no deleterious effects on the motility of ependymal cilia. 96 Therefore, the significant structural and functional differences in various types of motile cilia require rigorous functional and ultrastructural studies to definitively determine the role of ependymal cilia in hydrocephalic mutant mice. Nevertheless, we found strong evidence supporting a role for ciliary dysfunction in the pathogenesis of congenital hydrocephalus in 8 of 12 lines of genetically engineered mice (Ulk4, Nme5, Nme7, Kif27, Stk36, Dpcd, Ak7, and Ak8), each of which is discussed in detail below.
Ulk4 (aliases include A730098P15 and 4932415A06Rik): 28 ULK4 is a hypothetical protein kinase containing a protein kinase domain (Pfam accession No. PF00069), and its function has not been reported. The lesions noted in the nasal passages and brains of Ulk4 –/– mice are identical to those seen in many other mouse models of hydrocephalus with ciliary dyskinesia, strongly suggesting that Ulk4 is involved in the development and/or function of motile cilia in the upper respiratory tract and ependyma. The decreased or ineffective ciliary motility in mice with ciliary dyskinesia results in poor clearance of pathogens and secretions in the nasal passages and middle ear, which leads to bacterial colonization and suppurative inflammatory responses in these locations. As discussed previously, hydrocephalus in mice with ciliary dyskinesia is generally attributed to impaired ependymal flow.
Nme5 (aliases include Nm23-M5 and 1700019D05Rik): 9,27 NME5 is a putative nucleoside diphosphate kinase, which would make it a member of an enzyme family responsible for the synthesis of nucleoside triphosphates that is involved in numerous regulatory processes associated with proliferation, development, and differentiation. 45,68 Again, our findings of hydrocephalus, accumulation of nasal exudates, and abnormalities in spermatogenesis in Nme5 –/– mice strongly suggest that ciliary dysfunction is involved in the pathogenesis of these lesions. Interestingly, the ventricular dilatation/hydrocephalus and nasal exudates in Nme5 –/– mice were more severe than those seen in mice deficient for the related gene Nme7. However, Nme7 –/– mice displayed situs inversus, which is another sign suggestive of ciliary dysfunction affecting motile nodal cilia during development. 97 The absence of situs inversus in any Nme5 –/– mice indicates that the function of motile nodal cilia in these mice is unaffected, and it suggests that the axonemal defect may involve the structure or function of the central pair of microtubules rather than the outer 9 paired microtubules. In addition to our findings, there are reports in the literature suggesting a role for Nme5 in the development or function of motile cilia. These include the abundant expression of NME5 protein in tissues rich in highly ciliated cells, 64 the localization of NME5 adjacent to the central pair and outer doublets of axonemal microtubules in the flagella of spermatids and spermatozoa, 69 and the recent demonstration of a role for Nme5 in mediating the survival of round and elongated spermatids in mice. 17
Nme7 (aliases include Nm23-M7, Nm23-r7, and D5300-24H21Rik): 9,27 Until recently, 97 very little had been reported about the functions of this protein. Although the NDP kinase 7 encoded by Nme7 had not been previously linked to ciliary function in mammals, this kinase was reported to have significant homology to a flagellar protein (FAP67) in Chlamydomonas reinhardtii. 73 Based on our findings of situs inversus and hydrocephalus in Nme7 –/– mice, it seems likely that this kinase is involved in the biogenesis or function of motile cilia. Nevertheless, the absence of severe rhinitis/sinusitis and the normal male fertility observed in Nme7 –/– mice suggest that that the function and biogenesis of motile cilia/flagella in the respiratory epithelium and spermatozoa are less dependent on functional NDP kinase 7 than are the motile cilia of ependymal cells or in the embryonic node. The phenotypic differences between Nme5 –/– and Nme7 –/– mice illustrate the structural and functional variation in motile cilia at different locations (respiratory epithelium and embryonic node).
Kif27 (aliases for this gene include 4930517I18Rik and DKFZp434D0917): 53 Taken together, the lesions that we observed in Kif27 –/– mice are consistent with defective function of the ependymal and respiratory cilia. Decreased or ineffective ciliary motility results in decreased clearance of secretions in the nasal passages and middle ear and leads to bacterial colonization and suppurative inflammatory responses in these locations. As noted previously, hydrocephalus occurs frequently in mice with ciliary dyskinesis involving ependymal cilia. There is evidence in the literature supporting a role for Kif27 in the development or function of motile cilia. Kif27 is most abundantly expressed in tissues rich in highly ciliated cells, such as olfactory sensory neurons, and was therefore predicted to be important to cilia. 64 Although the function of KIF27 protein is not fully characterized, it is known to participate in the hedgehog signaling pathway in regulating highly conserved processes in development. 50–53 Interestingly, hedgehog transduction is mediated by a cytoplasmic signaling complex that includes the kinesin Costal 2 (homolog of Kif27 in mice and KIF27 in humans) and the serine–threonine kinase Fused (which is the homolog of Stk36 in mice). 53 Hedgehog signaling is essential for several aspects of embryogenesis, and in Drosophila, Hh transduction is mediated by a cytoplasmic signaling complex that includes the putative serine–threonine kinase Fused (Fu) and the kinesin Costal 2 (Cos2) 106 or the vertebrate homologs Kif27/Kif7. 15 KIF27 protein is a member of the kinesin family of ATP-powered enzymes that generate force along microtubules in Drosophila, therefore establishing the existence of fundamental mechanistic links between Hh signaling and ciliary function. 81 In planaria worms, a reduction in signal transduction proteins coded by cos2/kif27/kif7, fused, or iguana does not result in detectable Hh signaling defects but appears to be essential for planarian ciliogenesis. 81 Interestingly, the protein KIF27 was recently shown to physically interact with Fused (STK36), a critical protein in the construction of the central pair apparatus of motile 9+2 cilia in mice. 106 Taken together, these earlier reports, plus our findings of hydrocephalus and other lesions that are characteristic of ciliary dyskinesia, support an interactive role for both Kif27 and Stk36 in ciliogenesis.
Stk36 (aliases include FU, Fused, KIAA1278, MGC58023, mKIAA1278, B930045J24, and 1700112N14Rik): 65,106 As mentioned above, STK36 is a serine/threonine kinase and a homolog of Drosophila Fu protein (N-terminal homology only), which is in a pathway that promotes hedgehog-dependent gene transcription. The simultaneous occurrence of suppurative rhinosinusitis and hydrocephalus in our Stk36 null mice is strongly suggestive of an underlying functional defect in motile cilia. Our Stk36 –/– mice were similar to one previously reported line of Fu (Stk36) knockout mice in displaying severe growth retardation with suppurative rhinitis, hydrocephalus, and increased postnatal mortality 65 but differed markedly from yet another line of Stk36 (Fu) knockout mice in which mutant mice were stunted but showed no obvious pathology (including hydrocephalus), although most died before reaching 3 weeks of age. 15 The craniofacial defects we observed in some of our Stk36 null mice represent developmental defects in organs where hedgehog signaling is required (face, bones, etc); similar defects were not reported in either of the 2 previously reported lines of Stk36 null mice. 15,65 We are unable to explain the specific mechanisms responsible for the phenotypic differences noted in these 3 lines of Stk36 null mice but suspect that the different genetic backgrounds of mice used by the other groups may be involved.
Dpcd: 110 We previously reported the occurrence of situs inversus, hydrocephalus, and suppurative rhinosinusitis in Dpcd/Poll –/– mice. 97 All these findings are consistent with the defective biogenesis or function of motile cilia in multiple locations.
Ak7 (the alias for this gene is 4930502N02Rik): 30,66,71 Ak7 encodes adenylate kinase 7. Our Ak7 –/– knockout mice showed hydrocephalus, abnormal spermatogenesis, and mucus accumulation in nasal passages and paranasal sinuses, a phenotype that is consistent with primary ciliary dyskinesia. Our mice were virtually identical to another Ak7 –/– mouse model, 30 where electron microscopy confirmed the absence of many central microtubule doublets. This finding might also explain the normal situs in Ak7 –/– mice, as normal situs development depends on motile embryonic node cilia, which already lack central microtubules and would thus be less likely affected by central tubule defects.
Ak8 (aliases include 1190002A17Rik, MGC118311, MGC-130365, and RP23-362N19.5): 71 At the time that this report was originally submitted, there had been no reports in the scientific literature describing potential functions of the hypothetical protein encoded by this gene. Recently, however, this gene was identified as the newest member of the adenlylate kinase (AK) family, which comprises a group of enzymes that catalyze the nucleotide phosphoryl exchange reaction between adenosine monophosphate (AMP) and adenosine triphosphate. 71 AK7 and AK8 are cytosolic enzymes with high affinity for AMP and are very efficient in AMP phosphorylation. It has been postulated that AK7 and AK8 activities may be crucial to meeting the high-energy requirements of motile cilia. 71 Although mice deficient in either AK7 or AK8 each developed hydrocephalus, they differed markedly in disease severity and age of onset. Decreased viability was clearly evident in Ak7 –/–mice, which showed reduced number of homozygotes at time of weaning with very few mutant mice surviving beyond 3 weeks of age. The Ak7 –/– mice developed signs of severe hydrocephalus by 1 week of age, which included domed heads, small size, and stunted growth. In contrast, Ak8 –/– mice showed normal viability and did not develop lesions of hydrocephalus until after 2 weeks of age, which resulted in minimal doming of the skull. Moreover, Ak8 –/– mice displayed a normal behavioral phenotype in spite of developing hydrocephalus. Taken together, the relatively mild and later onset of hydrocephalus in Ak8 –/– mice suggests that ciliary dysfunction is less severe in these mice. The phenotype in Ak8 –/– mice suggests that they could serve as a model for late-onset communicating hydrocephalus.
The 4 remaining hydrocephalic mouse lines included in this report—RIKEN cDNA4930444A02, Celsr2, Mboat7, and Tg(FZD3)1Lex mice—do not have obvious ciliary lesions or involve known ciliary proteins. However, we cannot definitively exclude a pathogenetic role for ciliary dysfunction on the basis of our limited observations and what little is known of these genes. In fact, the cerebellar lesions observed in our RIKEN cDNA4930444A02 knockout mice show similarities to the human syndromes in which nonmotile primary cilia have been implicated. RIKEN cDNA 4930444A02 encodes a hypothetical protein that is an ortholog of a human hypothetical protein kinase-like protein named SgK196 (alias FLJ23356). To the best of our knowledge, there are no previous reports in the scientific literature that describe potential functions of the hypothetical protein encoded by this gene. A major feature of the mutant phenotype was the presence of misplaced neurons (heterotopia) in the cerebellum, hippocampus, and cerebral cortex, indicating that absence of this protein results in abnormal neuronal migration disorder in those tissues. It is now well known that nonmotile primary cilia are present on Purkinje cells and granule cell precursors and that these primary cilia play a role in normal cerebellar development. 16 Indeed, the presence of cerebellar abnormalities in several known ciliopathies suggests an important role for nonmotile primary cilia in normal development of the cerebellum. 4 , 16 , 37 , 63 , 67 , 86 Some of the best-known developmental disorders characterized by cerebellar malformations, which include Joubert syndrome, Meckel-Gruber syndrome, and Bardet-Biedl syndrome, have been shown to involve mutations in cilia-related genes. 16,63,67,86 It is clear that neuronal migration is a complex process involving cytoskeletal molecules controlling the initiation of migration, leading edge extension, and nucleokinesis; signaling molecules (the reelin pathway playing a central role) integrating external signals and linking them to the cytoskeleton; stop signals; and other molecular players, including neurotrophins, glutamate receptors, and peroxisome-derived factors (reviewed in Gressens 40 ).
Any of the many developmental processes that have been implicated in the pathogenesis of hydrocephalus could be involved in the Celsr2 –/–, Mboat7 –/–, and homozygous Tg(FZD3)1Lex mice, including unrecognized involvement in ciliogenesis. Other known hydrocephalus-associated genes include cytokines and growth factors or related molecules that are involved in cellular signal pathways expressed during early brain development. For example, mutations in the L1CAM gene, which encodes the neural cell adhesion protein L1, have been associated with X-linked hydrocephalus in humans, 105 and L1-deficient mutant mice develop severe hydrocephalus that does not involve either aqueduct stenosis or ultrastructural abnormalities of ependymal cells or cilia lining the lateral ventricles or the aqueduct. 82 Hydrocephalus can also result from mesenchymal cell abnormalities; transgenic mice expressing the Gi-coupled GPCR Ro1 receptor in astrocytes develop hydrocephalus, 90 and hydrocephalic Msx1 mutant mice have malformations in the posterior commissure/subcommissural organ and collapsed cerebral aqueducts. 31,79 It is possible that the underlying pathogenetic mechanism responsible for the development of hydrocephalus in Celsr2 –/–, Mboat7 –/–, and Tg(FZD3)1Lex mice might involve disruptions in neuronal migration and brain morphogenesis.
Celsr2 (aliases include EGFL2, mfmi1, KIAA0279, Flam2ingo1, and mKIAA0279): 33,34,44,93 A role for Celsr2 in regulating the growth of axons and dendrites of tightly packed neurons during brain development has been demonstrated. Recent studies indicate that the homologous transmembrane cadherins Celsr2 and Celsr3 regulate neurite growth in opposite ways, with Celsr2 enhancing neurite growth and Celsr3 suppressing it. 85 It also appears that Celsr2 may play a role in the migration of neurons to their final target area. In zebrafish, celsr2 in neuroepithelial cells was shown to regulate the polarity of neurons during development. 99 Conditional inactivation of Celsr3 demonstrated essential roles for the gene, in the neurons that project their axons to subcerebral targets such as the spinal cord, as well as in cells that guide projecting axons through the ventral forebrain. 113 We hypothesize that the high incidence of hydrocephalus in Celsr2–/– mice may result from abnormalities in brain morphogenesis that result in compression and blockage of the aqueduct of Sylvius.
Mboat7 (aliases include BB1, mBB1, Leng4, Lpiat, and 5730589L02Rik): 35 There are only a few published reports on the function of MBOAT7. In neutrophils, it has been shown to have a role in recycling arachidonate and regulating free arachidonic acid levels and leukotriene synthesis. 35 In Caenorhabditis elegans, the orthologous mboa-7 is also involved in arachidonate metabolism and is required for incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mutant worms deficient for mboa-7 exhibited larval arrest and egg-laying defects. 60 The normal appearance of cilia on respiratory and ependymal epithelium in Mboat7 –/– mice and the absence of any evidence of defective ciliary function (normal situs, normal fertility, and normal nasal passageways) suggest that ciliary dysfunction (if present at all) must be limited to ependymal cilia.
Fzd3 (aliases include Fz-3, Fz3, AU020229, and D930050A07Rik): 101 The mouse gene is frizzled (Drosophila) homolog 3 (Fzd3), which is orthologous to human FZD3. Fzd3 is a member of the frizzled gene family, which encodes 7-transmembrane domain proteins that are receptors for the Wingless family of proteins. 55 In the mouse, both Frizzled3 and Frizzled6 have been shown to control axonal growth and proper midbrain morphogenesis in the CNS. 88,102 In previous studies, our neonatal Fzd3 –/– mice had curly or looped tails (a sensitive indicator of neural tube defects) and typically died within 30 minutes of birth (data not shown). Our findings of early lethality and curly tails in Fzd3 –/– mice were similar to those reported previously in other Fzd3 –/– mice, 100–102 where targeted deletion of the mouse Fzd3 gene resulted in severe defects in several major axon tracts within the forebrain regions of the CNS. 101 Interestingly, the only grossly evident neuroanatomic defect exhibited by Fzd3 knockout embryos in this previous report was the marked enlargement of the lateral ventricles, which progressed in severity between days E15 and E18 and was attributed to shrinkage of the striatum and thinning of the cortex. 101 The CNS lesions that we observed in humanized Tg(FZD3)1Lex mice were mild in comparison to severe effects on midbrain morphogenesis and early lethality resulting from Fzd3 gene deletion in mice, and they suggest that the partial restoration of frizzled 3 activity in humanized Fzd3 mutant mice was sufficient to rescue mice from severe CNS lesions and early lethality. However, this rescue was not sufficient to prevent the development of hydrocephalus and relatively mild neurobehavioral changes. In our experience, the partial rescue of severe phenotypes in similarly “humanized” lines of mice is not unprecedented. 95 Taken together, these findings suggest that hydrocephalus in the humanized Tg(FZD3)1Lex mice could be due to aqueduct obstruction caused by defective development of midbrain structures. This Tg(FZD3)1Lex mouse line might prove useful in studies on the effects of frizzled 3 on brain development.
In conclusion, our findings in this survey of over 4650 different knockout mouse lines indicate that ciliary dyskinesia is likely the most common underlying pathogenetic mechanism responsible for the development of congenital hydrocephalus in mice, although a variety of other processes could be involved. Since the first reports of autosomal recessive hydrocephalus in mice in the 1930s, 18,19 many animal models of congenital hydrocephalus caused by genetic mutations have been reported. 10,75,78,107 A recent review listed over 40 mutants/loci and 9 specific genes associated with the development of congenital hydrocephalus in animal models, 111 and additional genes are being identified in knockout mice with increasing frequency. The high prevalence of familial patterns of inheritance for congenital hydrocephalus in humans 94 suggests that identification of genes responsible for development of hydrocephalus in mice may lead to the identification of homologous modifier genes and susceptibility alleles in humans. Even though the direct relevance of mouse models to hydrocephalus in humans remains uncertain, there is no doubt that genetically modified mouse models can add to our understanding of the cell signaling and developmental pathways involved in the pathogenesis of hydrocephalus.
Footnotes
Acknowledgements
We thank June Wingert, Ryan Vance, Kathy Henze, and Mary Thiel for their invaluable skills and efforts in providing necropsy and histology support. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
All authors were employees of Lexicon Pharmaceuticals Inc. the Woodlands, TX 73881.