
Abstract
Tubulin tyrosine ligase–like 1 (TTLL1) protein is a member of the tubulin tyrosine ligase superfamily of proteins that are involved in the posttranslational polyglutamylation of tubulin in axonemal microtubules within cilia and flagella. To investigate the physiological role of TTLL1, the authors generated mice with a gene trap mutation in the Ttll1 gene that provide confirmation in a mammalian model that polyglutamylation plays an important role in some ciliary and flagellar functions. For the first time, mice homozygous for the Ttll1 mutation exhibited accumulations of exudates in the nasal passages and sinuses, rhinosinusitis, otitis media, and male infertility. In homozygous mutant male mice, abnormal sperm morphology and function were characterized by shortened or absent flagella and immotility. Although homozygous mutant males were infertile, the females were fertile. These findings are consistent with a diagnosis of primary ciliary dyskinesia (PCD) resulting from ciliary dysfunction. They indicate that Ttll1 is essential for normal motility of respiratory cilia and the biogenesis and function of sperm flagella but that the defect does not result in the hydrocephalus or laterality defects often seen in other forms of PCD. The absence of early-onset lethal hydrocephalus in Ttll1-mutant mice may enable studies to evaluate the long-term effects of PCD in the respiratory system of mice. Although no mutations in the orthologous gene have been linked with PCD in humans, investigating the role of TTLL1 and polyglutamylation of tubulin in cilia and flagella should advance an understanding of the biogenesis and function of these organelles in mammals and have potential diagnostic and therapeutic applications.
Keywords
Abnormalities in the structure and/or function of motile and sensory cilia have been linked to a range of diseases now classified as ciliopathies. 33 Primary ciliary dyskinesia (PCD), formerly known as immotile cilia syndrome, comprises a group of rare, genetically heterogeneous but usually autosomal-recessive disorders that develop as a result of dysfunctional motile cilia and flagella. 3 Cilia and flagella depend on a tubulin-based cytoskeleton (axoneme) for structure and motility. The term PCD is used to describe genetic defects in cilia to differentiate them from the transient secondary ciliary dyskinesia that can follow epithelial injury based on viral infections 9 or exposure to pollutants. 8,42 In humans, the overall prevalence of PCD is around 1 in 20,000 to 30,000, 2 although much higher prevalence is reported in populations with increased levels of consanguinity. 39
Motile flagella and cilia normally create fluid flow over cell surfaces 52 or propel cells through a fluid medium. 43 Ciliated cells line the respiratory tract (including the sinuses and middle ear), the ventricular system of the brain, the female oviduct, and the male epididymis and vas deferens; thus, PCD is characterized by lesions in organs that depend on ciliary motility for proper function, such as the upper and lower respiratory tract, middle ear, reproductive organs, and brain. Cilia in the respiratory tract assist airway clearance of respiratory secretions, and because mucociliary clearance is impaired in PCD, 18 patients develop chronic otitis media, rhinitis, sinusitis, and recurrent infections of the lower respiratory tract that can result in irreversible bronchiectasis. 20,34
Other common signs of ciliary dyskinesia include hydrocephalus and laterality defects. 6 Impaired ependymal flow contributes to the development of hydrocephalus, whereas the randomization of left–right body asymmetry is due to dysfunctional nodal cilia during early embryogenesis. Laterality defects occur in nearly 50% of PCD patients, and they range from situs inversus totalis 38 to variable degrees of heterotaxy that are associated with complex heart defects. 29 In spermatozoa, flagellar defects can cause male infertility, whereas female fertility does not appear to be markedly impaired in PCD, despite the presence of ciliated epithelium of the oviduct. PCD has been associated with defects in a growing number of cilia-associated genes, and the complexity of ciliary structure and function is reflected by the many different clinical presentations associated with ciliary abnormalities. 51
Cilia and flagella are highly complex organelles made up of hundreds of different polypeptides that could be important for normal function. 26 A diverse array of genes has been implicated in the pathogenesis of PCD; however, much is still unknown about the biochemical and cellular processes responsible for ciliary and flagellar formation and function. A diverse array of genes has been implicated in PCD in mice, but in humans, mutations have so far been identified in only 8 genes in PCD (DNAI1, DNAH5, DNAH11, DNAI2, KTU, RSPH9, RSPH4A, and TZXNDC3). 31 For example, mutations in dynein genes cause PCD by producing axonemal defects, such as missing inner and/or outer dynein arms that impair the function of motile cilia. Mutations in the heavy (dynein axonemal heavy chain 5, or DNAH5) or intermediate (dynein axonemal intermediate chain 1, or DNAI1) chain dynein genes, which encode components of the ciliary outer dynein arms, are the most frequently identified genetic causes of PCD in humans. 31 However, the increased prevalence of chronic respiratory disease and bronchiectasis in patients with ciliopathies involving sensory cilia—including autosomal dominant or recessive polycystic kidney disease, Bardet-Biedl syndrome, and Alstrom syndrome—suggests that there can be some functional overlap in motile and immotile cilia. 31
Animal models for PCD have been identified in dogs, 13,14,59 cats, 47 pigs, 48 rats, 56 and mice. 32 In recent years, several different knockout mouse models have helped elucidate fundamental mechanisms and genes involved in the biogenesis and function of motile cilia; these models include mice deficient in Mdnah5, 23 Mdhc7, 37 Dpcd/poll, 64 Pcdp1, 30 Hydin, 10 Tektin-t, 55 Spag16, 65 Spag6, 50 Ak7, 17 and Nme7. 58 During the process of generating and analyzing more than 4,700 knockout mouse lines in a high-throughput mutagenesis and phenotyping process designed to characterize protein functions and identify novel drug targets, 62,63 we have identified additional knockout mice (with deletions in Nme5, Kif27, Stk36, Ulk4, and Ttll1) that have pathological phenotypes highly suggestive of PCD (unpublished observations). Because the ciliary genome is highly conserved from simple unicellular eukaryotes to mammals, these PCD-related genes in mice may prove useful in studying fundamental ciliary processes, and they may represent candidate genes for human PCD.
In this article, we report lesions in the respiratory tract and male reproductive tract of mutant mice deficient in the protein tubulin tyrosine ligase–like family, member 1 (TTLL1), which are similar to PCD lesions in mice. The tubulin tyrosine ligase–like (TTLL) proteins compose a large family of proteins whose members could catalyze ligations of diverse amino acids to tubulins or other protein substrates. 28 Polyglutamylation is a posttranslational modification in which polyglutamate side chains of variable lengths are added to target proteins such as tubulin and nucleosome assembly proteins. 12,44,45,49 The variable length of polyglutamate side chains may provide a mechanism for modulating protein–protein interactions, 27 and polyglutamylation of tubulin, the building block of microtubules, has been implicated in several functions of microtubules 27 and appears to play a role in axoneme motility. 19,22 Our findings strongly suggest that TTLL1 plays a role in establishing normal functions of specific cilia and flagella in mice. Further studies in this mouse model may enhance understanding of ciliary processes and development and lead to improved diagnosis, treatment, and outcomes in humans with PCD.
Materials and Methods
Mouse Production
We generated gene trap mutations in strain 129S5/SvEvBrd-derived embryonic stem cells as described. 1,62 The Ttll1-mutant line was generated by gene trapping using clone OST372941 from Lexicon’s OmniBank library. 1,62 This clone was selected for microinjection based on sequence similarity between the 3′ RACE tag obtained from this clone and the Ttll1 gene (accession No. NM_178869). Before injection, the exact location of the gene trap vector insertion within the Ttll1 gene was determined by inverse polymerase chain reaction (PCR) using primers complementary to the gene trap vector. 21 Gene disruption was confirmed by a direct analysis of gene expression using reverse transcription PCR. RNA was extracted from kidney and spleen tissue of wild-type and homozygous mutant mice with a bead homogenizer and RNAzol (Ambion, Austin, TX), according to manufacturer’s instructions. Reverse transcription was performed with SuperScript II (Invitrogen, Carlsbad, CA) and random hexamer primers, according to the manufacturer’s instructions. PCR amplification was performed with oligonucleotide primers for Ttll1 (5′-ACACTGCTTTGAATGCTACGGCTAC-3′ and 5′-CACTTGCAGTCGGGAATCTCGC-3′). The targeted embryonic stem cell clone was microinjected into C57BL/6-Tyrc-Brd (albino) blastocysts to generate chimeric animals that were bred to C57BL/6-Tyrc-Brd (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous gene-deficient mice. The F2 mutant mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (–/+) offspring of chimeric founder parents. Using the albino variant of C57BL/6 permits simple visual recognition of chimeric offspring. 66 Animal genotypes were determined by quantitative PCR as previously described. 21 In brief, DNA isolated from tail biopsy samples was assayed by quantitative PCR for the neo gene, which is present in the gene-trapping vector (VICTR 48, accession No. EU676804) used to generate the gene mutation described in this study.
Mouse Husbandry
Mice were housed in microisolator cages within a barrier facility, at 24°C, on a fixed 12-hour-light/dark cycle and were provided acidified water and Purina rodent chow (No. 5001, Purina, St. Louis, MO) ad libitum. Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines, which are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals. 36 Quarterly sentinel surveillance of the source colonies was conducted (Charles River Laboratories International, Inc, Wilmington, MA) and showed no evidence of rodent viruses, Mycoplasma, or Helicobacter spp.
Phenotypic Screen
Wild-type and Ttll1-mutant mice were subjected to a battery of phenotype screening exams as previously described. 5,62 Screening assays included behavioral tests (eg, circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), funduscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, hematology, peripheral blood fluorescence-activated cell sorter analysis, urinalysis, quantitative magnetic resonance, dual-energy x-ray absorptiometry scans, computerized axial tomography scans, microcomputed tomography scans, fertility testing, skin fibroblast proliferation assays, and pathology. Matings of wild-type and Ttll1-mutant mice were monitored daily for the presence of copulation plugs. Individual males were tested with up to 4 different female mice on separate occasions.
Histopathology
Immediately after euthanasia, knockout mice and age-matched normal control mice were fixed by cardiac perfusion with 10% neutral buffered formalin. Standard tissues examined included the following: heart, skeletal muscle, aorta, lung, kidney, renal lymph node, trachea, thyroid gland, parathyroid gland, mediastinal lymph node, adrenal gland, pituitary gland, thymus, salivary glands, cervical lymph node, esophagus, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, rectum, mesenteric lymph node, liver, gallbladder, spleen, brain, spinal cord, eyes, Harderian gland, urinary bladder, uterus, ovaries, fallopian tube, skin, mammary gland, bone, bone marrow, adipose tissue, blood, teeth, prostate gland, testes, epididymis, seminal vesicle, vas deferens, urethral glands, inguinal lymph node, inner ear, middle ear, nasal turbinates. Tissues were collected and immersed 10% neutral buffered formalin for an additional 48 hours, except for the eyes and testes, which were removed and fixed by immersion overnight at room temperature in Davidson’s (Poly Scientific, NY) and Bouin’s (Harleco EMD Chemicals, Gibbstown, NJ) fixatives, respectively. All tissues were embedded in paraffin, sectioned at 4 μm, mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA), and stained with HE for histopathologic examination.
Spermatozoa Motility and Structure
Sperm were collected by gently squeezing excised cauda epididymides of euthanized ENZ679T1 males to expel contents in collection media (KSOM, 370C, equilibrated in CO2). Collected sperm were incubated (370C, 5% CO2, 95% air) for 10 minutes and assessed for motility. Incubated sperm were pelleted (2 minutes at 10,000 × g) and suspended in formaldehyde (7.5% formaldehyde in phosphate buffered saline) for fixation (10 minutes). Fixed sperm were pelleted; fixative was discarded; and sperm were washed (0.1M ammonium acetate, pH 9). Supernatant was removed, leaving sufficient wash to gently suspend the pellet, which was spread onto slides and air-dried. Slides were stained with Coomassie solution (0.4% Coomassie G-250 in 3.5% [v/v] perchloric acid) for 10 minutes, rinsed with water until stained appeared blue, air-dried, and mounted with Permount.
Results
TTLL1 was disrupted by gene trapping as part of our large-scale mouse knockout study aimed at identifying the functions of 5,000 mouse genes within historically druggable gene classes. The gene trap mutation used in this study was localized to intron 10 of the Ttll1 gene and is predicted to truncate the protein at amino acid 326, within the second ATP-binding domain (Fig. 1 ). Previous studies of the related TTLL family member Tt116Ap in Tetrahymena have shown that mutation of this ATP-binding site renders the protein inactive. 28 Intercrossing heterozygote mice carrying the Ttll1 gene trap mutation generated wild-type, heterozygous, and homozygous F2 littermates in accordance with expected 1:2:1 Mendelian ratios (74:153:72; χ2 = 0.91), indicating normal embryonic and postnatal development. No significant differences between homozygous and wild-type littermates were noted in behavioral tests and almost all other screening tests. Male homozygous mice tended to be smaller than gender-matched wild-type littermates, and they exhibited decreased mean body weight and lean body mass when compared gender-matched (+/+) littermates and historical means (data not shown).
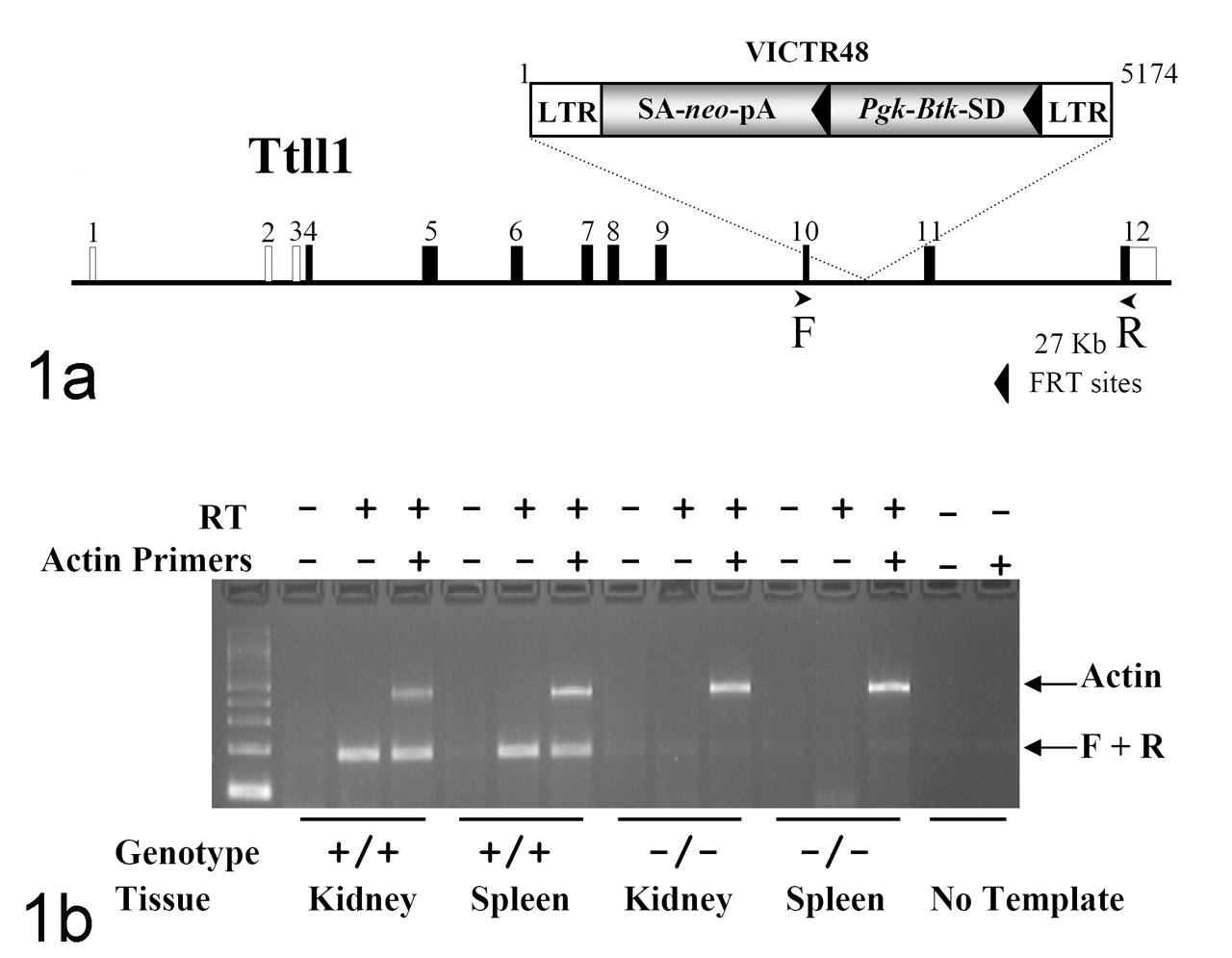
Generation of Ttll1-mutant mice. A, retroviral gene trap vector VICTR48 was used to produce clone OST372941, which contains an insertion within intron 10 of the Ttll1 gene. This mutation truncates the Ttll1 gene product after the seventh coding exon at amino acid 326 of protein. LTR, viral long terminal repeat; SA, splice acceptor sequence; neo, neomycin phosphotransferase gene; pA, polyadenylation sequence; Pgk, phosphoglycerate kinase–1 promoter; Btk-SD, Bruton tyrosine kinase splice donor sequence. B, reverse transcription polymerase chain reaction analysis of Ttll1 transcript. Endogenous Ttll1 transcript was detected in the kidney and spleen of wild-type (+/+) mice. No endogenous Ttll1 transcript was detected in homozygous (–/–) tissues. Primers F (5′-ACACTGCTTTGAATGCTACGGCTAC-3′) and R (5′-CACTTGCAGTCGGGAATCTCGC-3′) are complementary to Ttll1 exons 10 and 12, respectively, and amplify a product of 186 nucleotides. Analysis using primers complimentary to the mouse beta-actin gene (accession No. M12481) was performed in the same reaction as an internal amplification control.
The most consistent and notable abnormalities were observed in pathology, and the lesions in Ttll1-mutant mice are those commonly associated with PCD—the most prominent of which included chronic rhinosinusitis, otitis media, and male infertility. In marked contrast to findings in homozygous mice, no respiratory, reproductive, or other notable lesions were detected in either male or female heterozygous mice. In all Ttll1-mutant mice examined, there were accumulations of mucus and mucopurulent exudates in the nasal passageways and sinuses, which was frequently associated with chronic active inflammation in the nasal submucosa (Fig. 2 ). Similarly, the high prevalence of unilateral and bilateral suppurative otitis media in Ttll1-mutant mice is likely the result of impaired mucociliary function within the Eustachian tubes (Fig. 3 ). Histologically, normal-appearing cilia were present on respiratory epithelial cells lining the nasal passages and sinuses (Fig. 4 ), but the accumulation of exudates in the upper respiratory tract suggest impaired mucociliary clearance that predisposes the mice to chronic infections. Interestingly, there was no evidence of airway blockage or inflammation in the lower respiratory tract, and the lungs appeared to be completely normal (Fig. 5 ). Significantly, situs inversus was not detected in Ttll1-mutant mice; there was no evidence of dilation of the lateral and third ventricles of the brain; and cilia on ependymal cells were histologically unremarkable (Fig. 6 ).
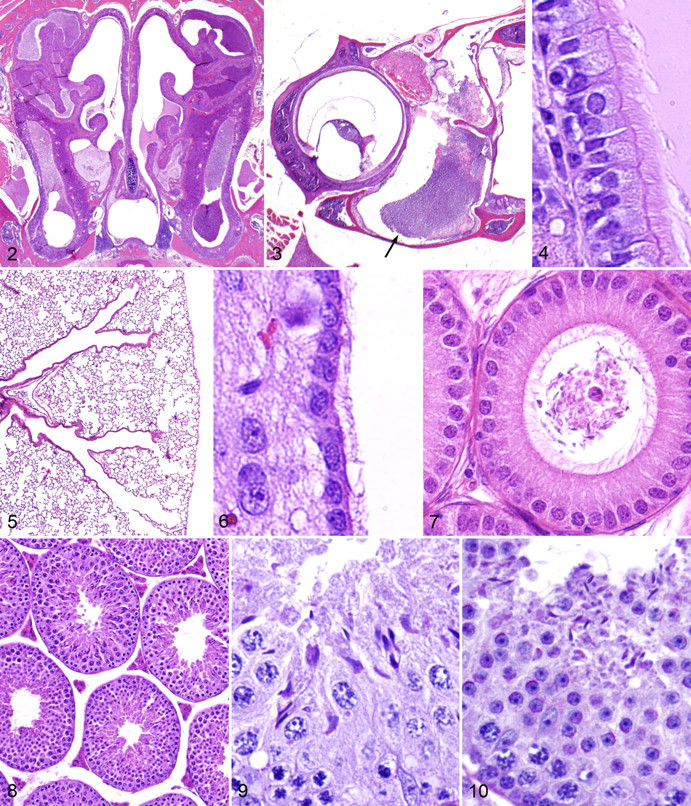

Heterozygous mice used for mouse production were fertile, but all homozygous male Ttll1-mutant mice were infertile. Histological examination showed abnormal development and maturation of spermatozoa in seminiferous tubules, and only small numbers of abnormal spermatozoa were present in the seminiferous tubules, efferent ducts, and epididymides of Ttll1-mutant mice (Fig. 7 ). Wild-type female mice that mated with homozygous male mice for extended periods showed vaginal plugs but did not become pregnant. The testes, seminal vesicles, and other reproductive organs of Ttll1-mutant male mice were grossly normal, and light microscopy analysis of histological sections of the testes showed normal architecture of the seminiferous tubules and interstitium (Fig. 8 ). Normal secondary sex differences were noted in the granular ducts of the submandibular salivary glands and parietal glomerular epithelium, suggesting normal testosterone production. However, there were striking differences between the mutant and wild-type sperm morphology and motility.
Histological analysis showed that although seemingly normal elongated spermatid heads were present in the seminiferous tubules and epididymis, the spermatozoa were characterized by thickened irregular midpieces, absent or markedly truncated flagella, and decapitation. No apparent defects were evident in spermatogonia during mitosis and meiosis (Fig. 9 ), and sperm heads were released into the lumen normally at spermiation, but flagellar loss appeared to take place before spermiation of elongate spermatids (Fig. 10 ). Histologically, normal spermatozoa were observed in the epididymis from wild-type and heterozygous mice. On wet mounts of fresh semen, normal spermatozoa in wild-type littermates displayed vigorous flagellar activity and progressive movement (Fig. 11 ). The spermatozoa obtained from mutant mice were severely reduced in number and immotile; morphologically, the spermatozoa in mutant mice were characterized by truncated or absent tails and swollen midpieces (Fig. 12 ). Although all Ttll1-mutant male mice were infertile, the apparently normal fertility of female Ttll1-mutant mice was demonstrated by productive breeding with wild-type mates; no differences in litter sizes or time to pregnancy were observed in several matings of Ttll1-mutant females (data not shown). Heterozygous male and female mice were all fertile and produced similar numbers of pups per pregnancy as wild-type littermates (data not shown).
Discussion
In humans, PCD and Kartagener syndrome are associated with recurrent respiratory infections, situs inversus, male infertility, and in some cases, hydrocephalus. In this study, we report lesions consistent with a diagnosis of PCD in mice having an inactivating insertional mutation in Ttll1 (Aliases for the mouse gene are 6330444E16Rik and AV014541). Ttll1-mutant mice developed chronic mucopurulent rhinitis, sinusitis, and otitis media, most likely as a result of impaired mucociliary clearance. Additional causes of sinusitis can be considered, such as decreased mucosal immunity and alteration of mucus composition, but these possibilities are not consistent with the demonstrated tubulin-modifying function of TTLL1.
Although some of the common signs of PCD (such as situs inversus and hydrocephalus) were not present in our Ttll1-mutant mice, the male infertility owing to evident flagellar defects strongly suggests that defective biogenesis and function of cilia and flagella are involved. It has long been known that structural defects in the microtubule-based axonemal structures of cilia and flagella are often involved in impaired ciliary/flagellar motility. Around 90% of individuals with PCD appear to have ultrastructural defects affecting the outer and/or inner dynein arms, which give cilia their motility, and almost 40% of these are associated with mutations in just 2 genes (DNAI1 and DNAH5) that encode for outer dynein arm proteins. Many mouse models of PCD have defects involving the inner and outer dynein arms in cilia. For example, mutant mice lacking a dynein chain protein (Mdnah5) have immotile cilia and develop chronic respiratory infections, hydrocephalus, and situs inversus. 23 On a C57BL/J genetic background, mice homozygous for the mutation of Pcdp1 (PCD protein 1) accumulate mucus in nasal passages and male infertility and often die perinatally from severe hydrocephalus. 30 Similarly, Tektin-t-deficient homozygous mutant males are infertile because of impaired motility of flagella as a result of disruption of dynein inner arm structures in the sperm flagella, although females are fully fertile. 55 Although male mice lacking the mouse dynein heavy chain 7 gene (Mdhc7), which encodes a component of the inner dynein arm, are viable and show no malformations consistent with PCD, they are nonetheless infertile owing to dramatically reduced directional motility of spermatozoa. 37 Our findings suggest that Ttll1-mutant mice have cilia in the upper respiratory tract with normal morphology but dysfunctional motility and severe structural defects in formation of spermatozoal flagella.
The basic axonemal structure of cilia and flagella consists of microtubules, which are in turn made up of α and β monomers of tubulin that form helical protofilaments. 54 The recently discovered polyglutamylases provide the first evidence of the importance of polyglutamylation in microtubule functions. Tubulin tyrosine ligase–like protein 1 (TTLL1; aliases include PGs3, C220rf7, HS323M22B) is involved in the polyglutamylation of α tubulin, a poorly understood but highly conserved posttranslational modification that is apparently required for efficient assembly and/or function of a subset of microtubule-based organelles. 60 In mouse brain, tubulin polyglutamylase was shown to contain 3 polypeptides, the smallest of which was named the polyglutamylase subunit 1 (PGs1, for phosphatidyl glycerophosphate synthase 1). PGs1 was shown to be encoded by the GTRGEO22 gene, 46 and it forms part of a 5-subunit multiprotein complex. 28 The enzymatic activity of this complex was attributed to PGs3 (now known as TTLL1), based on obvious structural similarities with a known protein, tubulin tyrosine ligase. 15
It is not yet known how microtubule glutamylation alters the properties of microtubules, but recent findings in animal models indicate that polyglutamylation of tubulin is required for normal ciliary motility and structure.
In zebrafish, fleer (flr) encodes a protein that is required for axonemal tubulin polyglutamylation. 41 Zebrafish flr mutants exhibit renal cysts, randomized left–right asymmetry, hydrocephalus, and retinal degeneration, suggesting a defect in ciliogenesis. Because fleer protein lacks a tubulin tyrosine ligase domain, 57 it is unlikely to be the catalytic subunit of tubulin polyglutamylase but instead may be involved in the regulation, localization, or transport of TTLL enzyme activity. 41 Mutation of Pgs1 (GTRGEO22) in ROSA22 mice has been associated with male sterility and neuronal defects. 7 Subsequent analysis of the these Pgs1-mutant mice has revealed a notable loss of polyglutamylation on α tubulin, 25 indicating that PGs1 is important for tubulin polyglutamylation. PGs1 lacks catalytic activity, but its localization at tubulin polyglutamylation sites in neurons, axonemes, and centrioles 46 suggests that it, like the fleer protein in zebrafish, may be involved in the regulation, localization, or transport of the larger polyglutamylase complex. Because PGs1 forms part of the multiprotein complex with TTLL1, it is not surprising that there are phenotypic similarities between the Pgs1-null mice and our Ttll1-deficient mice.
In both mutant mouse lines, male sterility is due to defective flagellar structure, whereas other aspects of spermatogenesis and maturation of the spermatid head appear to be normal to the point of spermiation. The normal mating behavior and normal sexual dimorphism that we observed in the salivary glands and kidneys suggest that testosterone production was adequate in Ttll1-mutant male mice, not unlike Pgs1-null males that show normal levels of circulating testosterone and normal mating behavior. However, female mice in both mutant lines were fertile, suggesting that ciliary function in oviducts is less critical to female fertility. These fertility findings are consistent with those in humans with PCD, where reduced fertility in men with PCD is attributed to dysmotility of spermatozoa and/or reduced sperm count, whereas lack of ciliary motility in the fallopian tubes of women does not appear to have a major effect on fertility. 4
Suppurative rhinosinusitis is the most characteristic and consistent finding in mouse models of PCD. Ciliary dysfunction reduces the ability of the mucociliary clearance mechanism to remove secretions and infectious agents from mucosal surfaces, resulting in accumulation of exudates and promoting secondary bacterial infections. We initially thought that the inflammatory nasal exudates could have contributed to retarded growth in TTLL1-deficient mice by affecting food intake; however, we observed decreased body weight in males only, although suppurative rhinosinusitis was equally severe in both genders. Similarly, male Pgs1-mutant mice are smaller than wild-type controls, but the reduced body weight and body fat were apparently not due to alterations in resting metabolic rate, decreased food intake, or anosmia. 7 Interestingly, no lesions of the upper respiratory tract were reported in the Pgs1-mutant mice, 7 suggesting that TTLL1 enzymatic polyglutamylation activity may be less dependent on the concurrent activity of Pgs1 in respiratory cilia than in sperm flagella. In humans, defects in structure and/or function of motile cilia disrupt mucociliary clearance, leading to recurrent respiratory tract infections in humans that often progress to chronic sinusitis or rhinitis and even permanent lung damage (bronchiectasis). Interestingly, mice appear to be resistant to PCD-associated pulmonary disease, and their respiratory tract lesions are restricted to the nasal passageways and sinuses.
In humans, impaired mucociliary clearance in bronchial airways can lead to chronic pulmonary infections and, eventually, severe bronchiectasis. In marked contrast, the absence of pulmonary lesions in Ttll1-mutant mice is typical in mouse models of PCD; we have not observed pulmonary lesions in any of the mutant mouse lines showing any combination of rhinosinusitis, hydrocephalus, situs inversus, and/or male infertility (unpublished observations).
Although pulmonary lesions are consistently absent in mutant mice with PCD, it appears that mice with PCD frequently develop hydrocephalus, with the dilatation of brain ventricles being attributed to disruption of the flow of cerebrospinal fluid normally generated by ciliated ependymal cells. 24 Mouse strain–dependent genetic background effects can influence the incidence of hydrocephalus in PCD mouse models. For example, Nm1054-mutant mice on a B6 background usually die within the first week of life because of severe hydrocephalus, whereas mutants on a 129 background develop either mild or no hydrocephalus, indicating the presence of varying genetic modifiers in the different mouse strains. 30 In any event, the absence of hydrocephalus in our Ttll1-mutant mice is an unusual and potentially valuable finding because studies of PCD in mouse models have been hampered by the high incidence of lethal hydrocephalus caused by disruption of ciliary genes in mice. A transgenic mouse with deceased mucociliary clearance but without hydrocephalus was recently developed to provide a viable mouse model for long-term studies of PCD. 40 As in other murine PCD models, these transgenic mice developed severe rhinosinusitis, demonstrating the importance of mucociliary clearance to the health of the upper airways. However, and consistent with findings in all other PCD models in mice, no evidence of lung disease was observed in these transgenic mice up to 11 months, suggesting that other factors are able to compensate for the lack of mucociliary clearance in the lower airways of mice. 40 The absence of discernable lesions in the central nervous system and normal neurobehavioral test results in Ttll1 null mice suggest that this enzyme is not essential for neuronal or axonal function.
Similarly, the absence of situs inversus in Ttll1-mutant mice indicates that TTLL1 is not required to establish normal structure or function of nodal cilia. Because a major difference between motile nodal cilia and other motile cilia is that they lack a central microtubule pair, we speculate that the TTLL1 deficiency has primary effects on the functions of the central microtubule functions. Interestingly, there is a report of a human family with a mutation affecting the central microtubule pair that lead to PCD without situs inversus. 53 Little is known about the mechanism through which interactions between radial spokes and the central pair complex regulate dynein activity, but it is clear that most mutations affecting central pair structure completely block motility. 35,61 Taken together, these findings provide evidence that polyglutamylation may be critically important in central microtubule function.
The presence of histologically normal respiratory cilia and the concurrent virtual absence of sperm flagella in these Ttll1-mutant mice indicates significant differences in the way that TTLL1 affects the biogenesis and function of these related organelles, even though they share a common axonemal structure. It is not unprecedented that mutant mice with severe defects in the structure or function of flagella would also have structurally normal motile cilia. Although Pgs1-mutant mice have severely truncated sperm flagella, no abnormalities in axonemal structures of motile cilia or lesions attributable to lesions in respiratory cilia were reported. Similarly, Pcdp1-null mice showed complete disruption of sperm flagella but normal ciliary ultrastructure. 30 The sperm flagellum has a complex cytoskeletal structure, with the microtubular axonemes being surrounded by outer dense fibers, which are in turn encompassed by a mitochondrial sheath in the midpiece and a fibrous sheath in the principal piece (reviewed in Dutcher 11 ). Several genes that affect the development and function of the spermatid flagellum have been in knockout mice in recent years (reviewed in Escalier 16 ), and Ttll1 and Pgs1 appear to have similar effects. Ultrastructural studies on Ttll1-mutant sperm could help determine if deficiencies in TTLL1 and Pgs1 have similar effects on flagellar structure.
In conclusion, PCD is a clinically and genetically heterogeneous syndrome, and current diagnostic limitations argue for a more comprehensive effort to define disease-causing defects. The identification of disease-causing mutations and genetic testing should also provide a clearer understanding of the clinical spectrum of respiratory tract involvement in this disease. Many candidate genes important for ciliary function that could be associated with PCD have been identified by studies of primitive protozoans. Here, we demonstrate the utility of genetic loss-of-function studies in mice to identify novel genes required for normal ciliary function that could be candidate genes for genetic analysis in human PCD. Our findings in Ttll1-mutant mice suggest that TTLL1 is essential for normal motility of respiratory ciliary motility and for the biogenesis and structural integrity of sperm flagella. The findings provide confirmation in a mammalian model that polyglutamylation plays an important role in cilia and flagella. 19,22 Ttll1-mutant mice may provide a particularly useful model for studying PCD and male infertility because they do not develop early onset lethal hydrocephalus. Although no mutations in the orthologous gene have been linked with PCD in humans, investigating the role of TTLL1 in the polyglutamylation of tubulin in cilia and flagella may advance our understanding of the biogenesis and function of these important organelles and have potential diagnostic and therapeutic applications in the future.
Footnotes
Acknowledgements
We thank Mary Thiel, Kathy Henze, and Ryan Vance for histology support on this project. This research received no grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
Financial support for this research was provided by Lexicon Pharmaceuticals Inc, The Woodlands, TX.